

content
1、背景介绍[0]
2、生成.fasta代码[0]
3、批量化运行RoseTTAFold并输出相应的特征[0]
(1)下载RoseTTAFold安装包[0]
(2)创建conda环境[0]
(3)下载预训练权重[0]
(4)下载第三方安装包[0]
(5)下载相应的数据库:或者直接使用迅雷下载[0]
(6)测试[0]
4、批量化运行RoseTTAFold[0]
(1)修改predict_e2e.py[0]
(2)修改make_msa.sh[0]
(3)修改run_e2e_ver.sh[0]
(4)批量化运行并输出结果[0]
5、AlphaFold2原理介绍和安装[0]
1、背景介绍
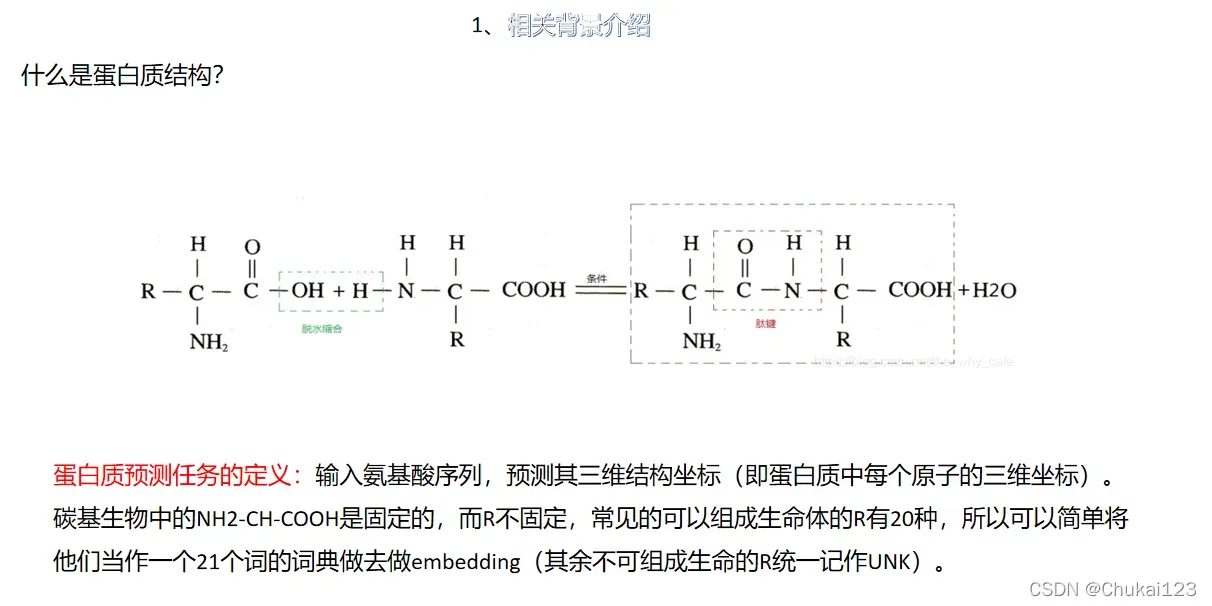



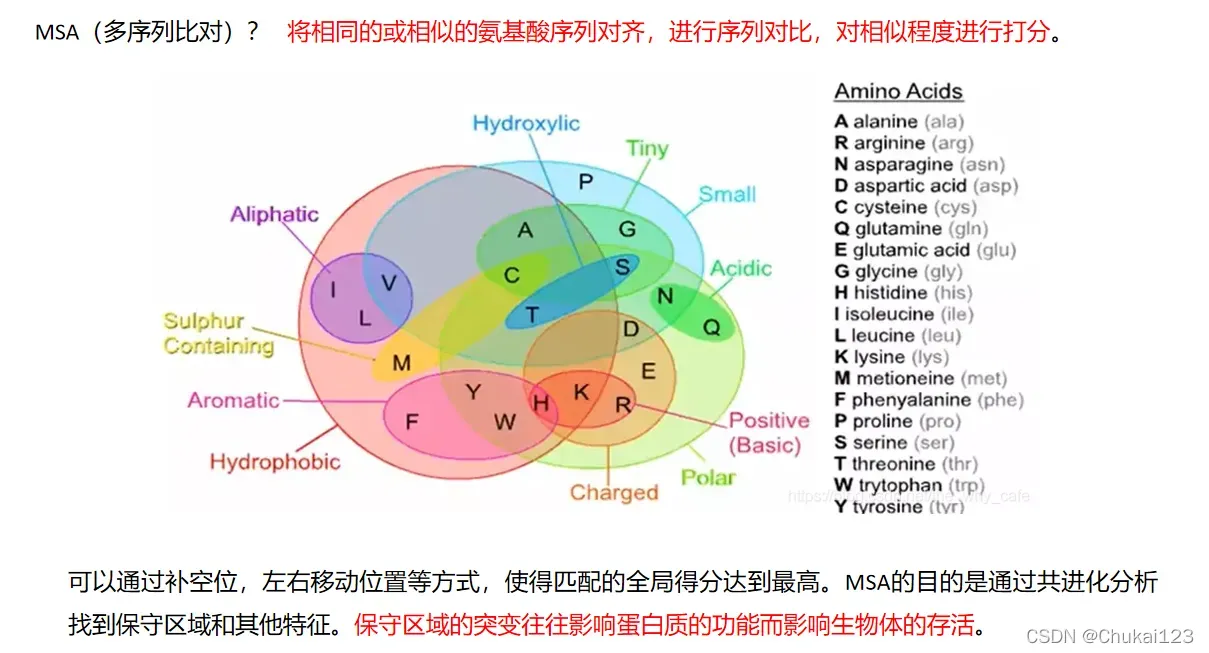



.fasta文件或者.fa文件(两个是一样的),这里是只是输出下面这种形式,在RoseTTAFold和alphafold中的输入形式是一样的
>T1078 Tsp1, Trichoderma virens, 138 residues|
MAAPTPADKSMMAAVPEWTITNLKRVCNAGNTSCTWTFGVDTHLATATSCTYVVKANANASQASGGPVTCGPYTITSSWSGQFGPNNGFTTFAVTDFSKKLIVWPAYTDVQVQAGKVVSPNQSYAPANLPLEHHHHHH
2、生成.fasta代码
input_file的格式:
| HLA | sequence |
|---|---|
| T1078 | DKSMMAAVPEWTITNLKRVCNAGNTSCTWTFGVDTHLA…… |
"""
author:chukai
time:2022/04/25
content:输入一个sequence文件,输出对应的.fasta文件
usage:
1、mkdir test
2、python generate_fasta.py -i 'XXXXXX.csv' -o test/
3、rm -rf test
"""
import pandas as pd
import argparse
def generate(path,output_path):
data = pd.read_csv(path)
for i in range(len(data)):
fastafile = []
MHC = data['HLA'][i].replace(':','')
length = len(data['sequence'][i])
fastafile.append('>' + MHC + "\t" + str(length))
fastafile.append(data['sequence'][i])
pd.DataFrame(fastafile).to_csv(output_path + MHC +'.fa', index=0, header=0)
if __name__ == '__main__':
parser = argparse.ArgumentParser(description='输入sequence获得.fasta文件')
parser.add_argument('-i', help='please input an file eg xxx.csv',default='xxxx.csv')
parser.add_argument('-o', help='the output of .fasta file',default='/home/chuk/xxxxx/xxxxx/')
args = parser.parse_args()
input = args.i
output = args.o
generate(input,output)测试:在Linux中执行下面指令即可获得批量的.fasta文件
mkdir test
python generate_fasta.py -i 'xxxxxx.csv' -o test/
rm -rf test/3、批量化运行RoseTTAFold并输出相应的特征


(1)下载RoseTTAFold安装包
git clone https://github.com/RosettaCommons/RoseTTAFold.git
cd RoseTTAFold(2)创建conda环境
# create conda environment for RoseTTAFold
# If your NVIDIA driver compatible with cuda11
conda env create -f RoseTTAFold-linux.yml
# If not (but compatible with cuda10)
conda env create -f RoseTTAFold-linux-cu101.yml
# create conda environment for pyRosetta folding & running DeepAccNet
conda env create -f folding-linux.yml(3)下载预训练权重
wget https://files.ipd.uw.edu/pub/RoseTTAFold/weights.tar.gz
tar xfz weights.tar.gz(4)下载第三方安装包
./install_dependencies.sh(5)下载相应的数据库:或者直接使用迅雷下载
# uniref30 [46G]
wget http://wwwuser.gwdg.de/~compbiol/uniclust/2020_06/UniRef30_2020_06_hhsuite.tar.gz
mkdir -p UniRef30_2020_06
tar xfz UniRef30_2020_06_hhsuite.tar.gz -C ./UniRef30_2020_06
# BFD [272G]
wget https://bfd.mmseqs.com/bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt.tar.gz
mkdir -p bfd
tar xfz bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt.tar.gz -C ./bfd
# structure templates (including *_a3m.ffdata, *_a3m.ffindex) [over 100G]
wget https://files.ipd.uw.edu/pub/RoseTTAFold/pdb100_2021Mar03.tar.gz
tar xfz pdb100_2021Mar03.tar.gz
# for CASP14 benchmarks, we used this one: https://files.ipd.uw.edu/pub/RoseTTAFold/pdb100_2020Mar11.tar.gz(6)测试
# For monomer structure prediction
cd example
../run_[pyrosetta, e2e]_ver.sh input.fa .
# For complex modeling
# please see README file under example/complex_modeling/README for details.
python network/predict_complex.py -i paired.a3m -o complex -Ls 218 310
# For PPI screening using faster 2-track version (example input and output are at example/complex_2track)
python network_2track/predict_msa.py -msa [paired MSA file in a3m format] -npz [output npz file name] -L1 [Length of first chain]
e.g. python network_2track/predict_msa.py -msa input.a3m -npz complex.npz -L1 2184、批量化运行RoseTTAFold
(1)修改predict_e2e.py
cd RoseTTAFold-main/network
vim predict_e2e.pyimport sys, os
# os.environ["CUDA_VISIBLE_DEVICES"] = "0,1"
import time
import numpy as np
import torch
import torch.nn as nn
from torch.utils import data
from parsers import parse_a3m, read_templates
from RoseTTAFoldModel import RoseTTAFoldModule_e2e
import util
from collections import namedtuple
from ffindex import *
from kinematics import xyz_to_c6d, c6d_to_bins2, xyz_to_t2d
from trFold import TRFold
script_dir = '/'.join(os.path.dirname(os.path.realpath(__file__)).split('/')[:-1])
NBIN = [37, 37, 37, 19]
MODEL_PARAM ={
"n_module" : 8,
"n_module_str" : 4,
"n_module_ref" : 4,
"n_layer" : 1,
"d_msa" : 384 ,
"d_pair" : 288,
"d_templ" : 64,
"n_head_msa" : 12,
"n_head_pair" : 8,
"n_head_templ" : 4,
"d_hidden" : 64,
"r_ff" : 4,
"n_resblock" : 1,
"p_drop" : 0.0,
"use_templ" : True,
"performer_N_opts": {"nb_features": 64},
"performer_L_opts": {"nb_features": 64}
}
SE3_param = {
"num_layers" : 2,
"num_channels" : 16,
"num_degrees" : 2,
"l0_in_features": 32,
"l0_out_features": 8,
"l1_in_features": 3,
"l1_out_features": 3,
"num_edge_features": 32,
"div": 2,
"n_heads": 4
}
REF_param = {
"num_layers" : 3,
"num_channels" : 32,
"num_degrees" : 3,
"l0_in_features": 32,
"l0_out_features": 8,
"l1_in_features": 3,
"l1_out_features": 3,
"num_edge_features": 32,
"div": 4,
"n_heads": 4
}
MODEL_PARAM['SE3_param'] = SE3_param
MODEL_PARAM['REF_param'] = REF_param
# params for the folding protocol
fold_params = {
"SG7" : np.array([[[-2,3,6,7,6,3,-2]]])/21,
"SG9" : np.array([[[-21,14,39,54,59,54,39,14,-21]]])/231,
"DCUT" : 19.5,
"ALPHA" : 1.57,
# TODO: add Cb to the motif
"NCAC" : np.array([[-0.676, -1.294, 0. ],
[ 0. , 0. , 0. ],
[ 1.5 , -0.174, 0. ]], dtype=np.float32),
"CLASH" : 2.0,
"PCUT" : 0.5,
"DSTEP" : 0.5,
"ASTEP" : np.deg2rad(10.0),
"XYZRAD" : 7.5,
"WANG" : 0.1,
"WCST" : 0.1
}
fold_params["SG"] = fold_params["SG9"]
class Predictor():
def __init__(self, model_dir=None, use_cpu=False):
if model_dir == None:
self.model_dir = "%s/models"%(os.path.dirname(os.path.realpath(__file__)))
else:
self.model_dir = model_dir
#
# define model name
self.model_name = "RoseTTAFold"
# if torch.cuda.is_available() and (not use_cpu):
# self.device = torch.device("cuda")
# else:
# self.device = torch.device("cpu")
self.device = torch.device("cuda:0")
self.active_fn = nn.Softmax(dim=1)
# define model & load model
self.model = RoseTTAFoldModule_e2e(**MODEL_PARAM).to(self.device)
def load_model(self, model_name, suffix='e2e'):
chk_fn = "%s/%s_%s.pt"%(self.model_dir, model_name, suffix)
if not os.path.exists(chk_fn):
return False
checkpoint = torch.load(chk_fn, map_location=self.device)
self.model.load_state_dict(checkpoint['model_state_dict'], strict=True)
return True
# 修改位置1
def predict(self, a3m_fn, out_prefix,inputt1d,inputt2d,count1d,count2d,temp1d,temp2d,hhr_fn=None, atab_fn=None,window=150, shift=75):
msa = parse_a3m(a3m_fn)
N, L = msa.shape
#
if hhr_fn != None:
xyz_t, t1d, t0d = read_templates(L, ffdb, hhr_fn, atab_fn, n_templ=10)
else:
xyz_t = torch.full((1, L, 3, 3), np.nan).float()
t1d = torch.zeros((1, L, 3)).float()
t0d = torch.zeros((1,3)).float()
#
msa = torch.tensor(msa).long().view(1, -1, L)
idx_pdb = torch.arange(L).long().view(1, L)
seq = msa[:,0]
#
# template features
xyz_t = xyz_t.float().unsqueeze(0)
t1d = t1d.float().unsqueeze(0)
t0d = t0d.float().unsqueeze(0)
t2d = xyz_to_t2d(xyz_t, t0d)
could_load = self.load_model(self.model_name, suffix="e2e")
if not could_load:
print ("ERROR: failed to load model")
sys.exit()
self.model.eval()
with torch.no_grad():
# do cropped prediction if protein is too big
if L > window*2:
prob_s = [np.zeros((L,L,NBIN[i]), dtype=np.float32) for i in range(4)]
count_1d = np.zeros((L,), dtype=np.float32)
count_2d = np.zeros((L,L), dtype=np.float32)
node_s = np.zeros((L,MODEL_PARAM['d_msa']), dtype=np.float32)
#
grids = np.arange(0, L-window+shift, shift)
ngrids = grids.shape[0]
print("ngrid: ", ngrids)
print("grids: ", grids)
print("windows: ", window)
for i in range(ngrids):
for j in range(i, ngrids):
start_1 = grids[i]
end_1 = min(grids[i]+window, L)
start_2 = grids[j]
end_2 = min(grids[j]+window, L)
sel = np.zeros((L)).astype(np.bool)
sel[start_1:end_1] = True
sel[start_2:end_2] = True
input_msa = msa[:,:,sel]
mask = torch.sum(input_msa==20, dim=-1) < 0.5*sel.sum() # remove too gappy sequences
input_msa = input_msa[mask].unsqueeze(0)
input_msa = input_msa[:,:1000].to(self.device)
input_idx = idx_pdb[:,sel].to(self.device)
input_seq = input_msa[:,0].to(self.device)
#
# Select template
input_t1d = t1d[:,:,sel].to(self.device) # (B, T, L, 3)
input_t2d = t2d[:,:,sel][:,:,:,sel].to(self.device)
np.savez_compressed("%s.npz"%(inputt1d),input_t1d.cpu())
np.savez_compressed("%s.npz"%(inputt2d),input_t2d.cpu())
#
print ("running crop: %d-%d/%d-%d"%(start_1, end_1, start_2, end_2), input_msa.shape)
with torch.cuda.amp.autocast():
logit_s, node, init_crds, pred_lddt = self.model(input_msa, input_seq, input_idx, t1d=input_t1d, t2d=input_t2d, return_raw=True)
#
# Not sure How can we merge init_crds.....
sub_idx = input_idx[0].cpu()
sub_idx_2d = np.ix_(sub_idx, sub_idx)
count_2d[sub_idx_2d] += 1.0
count_1d[sub_idx] += 1.0
np.savez_compressed("%s.npz"%(count2d),count_2d)
np.savez_compressed("%s.npz"%(count1d),count_1d)
node_s[sub_idx] += node[0].cpu().numpy()
for i_logit, logit in enumerate(logit_s):
prob = self.active_fn(logit.float()) # calculate distogram
prob = prob.squeeze(0).permute(1,2,0).cpu().numpy()
prob_s[i_logit][sub_idx_2d] += prob
del logit_s, node
#
# combine all crops
for i in range(4):
prob_s[i] = prob_s[i] / count_2d[:,:,None]
prob_in = np.concatenate(prob_s, axis=-1)
node_s = node_s / count_1d[:, None]
#
# Do iterative refinement using SE(3)-Transformers
# clear cache memory
torch.cuda.empty_cache()
#
node_s = torch.tensor(node_s).to(self.device).unsqueeze(0)
seq = msa[:,0].to(self.device)
idx_pdb = idx_pdb.to(self.device)
prob_in = torch.tensor(prob_in).to(self.device).unsqueeze(0)
with torch.cuda.amp.autocast():
xyz, lddt = self.model(node_s, seq, idx_pdb, prob_s=prob_in, refine_only=True)
else:
msa = msa[:,:1000].to(self.device)
seq = msa[:,0]
idx_pdb = idx_pdb.to(self.device)
t1d = t1d[:,:10].to(self.device)
t2d = t2d[:,:10].to(self.device)
# 修改位置2
np.savez_compressed("%s.npz"%(temp1d),t1d.cpu())
np.savez_compressed("%s.npz"%(temp2d),t2d.cpu())
with torch.cuda.amp.autocast():
logit_s, _, xyz, lddt = self.model(msa, seq, idx_pdb, t1d=t1d, t2d=t2d)
prob_s = list()
for logit in logit_s:
prob = self.active_fn(logit.float()) # distogram
prob = prob.reshape(-1, L, L).permute(1,2,0).cpu().numpy()
prob_s.append(prob)
np.savez_compressed("%s.npz"%(out_prefix), dist=prob_s[0].astype(np.float16), \
omega=prob_s[1].astype(np.float16),\
theta=prob_s[2].astype(np.float16),\
phi=prob_s[3].astype(np.float16))
self.write_pdb(seq[0], xyz[0], idx_pdb[0], Bfacts=lddt[0], prefix="%s_init"%(out_prefix))
# run TRFold
prob_trF = list()
for prob in prob_s:
prob = torch.tensor(prob).permute(2,0,1).to(self.device)
prob += 1e-8
prob = prob / torch.sum(prob, dim=0)[None]
prob_trF.append(prob)
xyz = xyz[0, :, 1]
TRF = TRFold(prob_trF, fold_params)
xyz = TRF.fold(xyz, batch=15, lr=0.1, nsteps=200)
xyz = xyz.detach().cpu().numpy()
# add O and Cb
N = xyz[:,0,:]
CA = xyz[:,1,:]
C = xyz[:,2,:]
O = self.extend(np.roll(N, -1, axis=0), CA, C, 1.231, 2.108, -3.142)
xyz = np.concatenate((xyz, O[:,None,:]), axis=1)
self.write_pdb(seq[0], xyz, idx_pdb[0], Bfacts=lddt[0], prefix=out_prefix)
def extend(self, a,b,c, L,A,D):
'''
input: 3 coords (a,b,c), (L)ength, (A)ngle, and (D)ihedral
output: 4th coord
'''
N = lambda x: x/np.sqrt(np.square(x).sum(-1,keepdims=True) + 1e-8)
bc = N(b-c)
n = N(np.cross(b-a, bc))
m = [bc,np.cross(n,bc),n]
d = [L*np.cos(A), L*np.sin(A)*np.cos(D), -L*np.sin(A)*np.sin(D)]
return c + sum([m*d for m,d in zip(m,d)])
def write_pdb(self, seq, atoms, idx, Bfacts=None, prefix=None):
L = len(seq)
filename = "%s.pdb"%prefix
ctr = 1
with open(filename, 'wt') as f:
if Bfacts == None:
Bfacts = np.zeros(L)
else:
Bfacts = torch.clamp( Bfacts, 0, 1)
for i,s in enumerate(seq):
if (len(atoms.shape)==2):
f.write ("%-6s%5s %4s %3s %s%4d %8.3f%8.3f%8.3f%6.2f%6.2f\n"%(
"ATOM", ctr, " CA ", util.num2aa[s],
"A", idx[i]+1, atoms[i,0], atoms[i,1], atoms[i,2],
1.0, Bfacts[i] ) )
ctr += 1
elif atoms.shape[1]==3:
for j,atm_j in enumerate((" N "," CA "," C ")):
f.write ("%-6s%5s %4s %3s %s%4d %8.3f%8.3f%8.3f%6.2f%6.2f\n"%(
"ATOM", ctr, atm_j, util.num2aa[s],
"A", idx[i]+1, atoms[i,j,0], atoms[i,j,1], atoms[i,j,2],
1.0, Bfacts[i] ) )
ctr += 1
elif atoms.shape[1]==4:
for j,atm_j in enumerate((" N "," CA "," C ", " O ")):
f.write ("%-6s%5s %4s %3s %s%4d %8.3f%8.3f%8.3f%6.2f%6.2f\n"%(
"ATOM", ctr, atm_j, util.num2aa[s],
"A", idx[i]+1, atoms[i,j,0], atoms[i,j,1], atoms[i,j,2],
1.0, Bfacts[i] ) )
ctr += 1
# 修改位置3
def get_args():
import argparse
parser = argparse.ArgumentParser()
parser.add_argument("-m", dest="model_dir", default="%s/weights"%(script_dir),
help="Path to pre-trained network weights [%s/weights]"%script_dir)
parser.add_argument("-i", dest="a3m_fn", required=True,
help="Input multiple sequence alignments (in a3m format)")
parser.add_argument("-o", dest="out_prefix", required=True,
help="Prefix for output file. The output files will be [out_prefix].npz and [out_prefix].pdb")
parser.add_argument("--db", default="/NewWorkSpace/rosettafold/pdb100_2021Mar03/pdb100_2021Mar03",
help="Path to template database [%s/pdb100_2021Mar03]")
parser.add_argument("--cpu", dest='use_cpu', default=False, action='store_true')
parser.add_argument("--input_t1d",dest='inputt1d',required=True,
help="save the input_t1d of input sequence after model iteration")
parser.add_argument("--input_t2d",dest='inputt2d',required=True,
help="save the input_t2d of input sequence after model iteration")
parser.add_argument("--count_1d",dest='count1d',required=True,
help="save the count_1d of input sequence after model iteration")
parser.add_argument("--count_2d",dest='count2d',required=True,
help="save the count_2d of input sequence after model iteration")
parser.add_argument("--t1d",dest='temp1d',required=True,
help="save the t1d of input sequence after model iteration")
parser.add_argument("--t2d",dest='temp2d',required=True,
help="save the t2d of input sequence after model iteration")
parser.add_argument("--hhr", default=None,
help="HHsearch output file (hhr file). If not provided, zero matrices will be given as templates")
parser.add_argument("--atab", default=None,
help="HHsearch output file (atab file)")
args = parser.parse_args()
return args
if __name__ == "__main__":
args = get_args()
FFDB=args.db
FFindexDB = namedtuple("FFindexDB", "index, data")
ffdb = FFindexDB(read_index(FFDB+'_pdb.ffindex'),
read_data(FFDB+'_pdb.ffdata'))
if not os.path.exists("%s.npz"%args.out_prefix):
pred = Predictor(model_dir=args.model_dir, use_cpu=args.use_cpu)
# add new args for inter_feature
# 修改位置4
pred.predict(args.a3m_fn, args.out_prefix,args.inputt1d,args.inputt2d,args.count1d,args.count2d,args.temp1d,args.temp2d,args.hhr, args.atab)
(2)修改make_msa.sh
修改数据库的地址:path
#!/bin/bash
# make the script stop when error (non-true exit code) is occured
set -e
# inputs
in_fasta="$1"
out_dir="$2"
# resource
CPU="$3"
MEM="$4"
path=/NewWorkSpace/rosettafold
# sequence databases
declare -a DATABASES=( \
"$path/UniRef30_2020_06/UniRef30_2020_06" \
"$path/bfd/bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt")
# setup hhblits command
HHBLITS="hhblits -o /dev/null -mact 0.35 -maxfilt 100000000 -neffmax 20 -cov 25 -cpu $CPU -nodiff -realign_max 100000000 -maxseq 1000000 -maxmem $MEM -n 4"
echo $HHBLITS
tmp_dir="$out_dir/hhblits"
mkdir -p $tmp_dir
out_prefix="$out_dir/t000_"
# perform iterative searches
prev_a3m="$in_fasta"
for (( i=0; i<${#DATABASES[@]}; i++ ))
do
for e in 1e-30 1e-10 1e-6 1e-3
do
echo db:$i e:$e
$HHBLITS -i $prev_a3m -oa3m $tmp_dir/t000_.db$i.$e.a3m -e $e -v 0 -d ${DATABASES[$i]}
hhfilter -id 90 -cov 75 -i $tmp_dir/t000_.db$i.$e.a3m -o $tmp_dir/t000_.db$i.$e.id90cov75.a3m
prev_a3m="$tmp_dir/t000_.db$i.$e.id90cov50.a3m"
n75=`grep -c "^>" $tmp_dir/t000_.db$i.$e.id90cov75.a3m`
echo " -- cov75 results: $n75"
if (($n75>2000))
then
cp $tmp_dir/t000_.db$i.$e.id90cov75.a3m ${out_prefix}.msa0.a3m
break 2
fi
hhfilter -id 90 -cov 50 -i $tmp_dir/t000_.db$i.$e.a3m -o $tmp_dir/t000_.db$i.$e.id90cov50.a3m
n50=`grep -c "^>" $tmp_dir/t000_.db$i.$e.id90cov50.a3m`
echo " -- cov50 results: $n50"
if (($n50>4000))
then
cp $tmp_dir/t000_.db$i.$e.id90cov50.a3m ${out_prefix}.msa0.a3m
break 2
fi
done
done
if [ ! -s ${out_prefix}.msa0.a3m ]
then
cp $prev_a3m ${out_prefix}.msa0.a3m
fi(3)修改run_e2e_ver.sh
配置数据库地址和输出文件
#!/bin/bash
# make the script stop when error (non-true exit code) is occured
set -e
############################################################
# >>> conda initialize >>>
# !! Contents within this block are managed by 'conda init' !!
__conda_setup="$('conda' 'shell.bash' 'hook' 2> /dev/null)"
eval "$__conda_setup"
unset __conda_setup
# <<< conda initialize <<<
############################################################
SCRIPT=`realpath -s $0`
export PIPEDIR=`dirname $SCRIPT`
CPU="8" # number of CPUs to use
MEM="64" # max memory (in GB)
# Inputs:
#####---------------------------------------------------------
#
# 后续调整的思路:
# dir=/WorkSpace/chuk/pdb100_2021Mar03
#
# for file in $dir/*; do
# echo $file # 输出目录下的每个文件
# done
#
#####---------------------------------------------------------
IN="$1" # input.fasta
WDIR=`realpath -s $2` # working folder
LEN=`tail -n1 $IN | wc -m`
mkdir -p $WDIR/log
conda activate RoseTTAFold
############################################################
# 1. generate MSAs
############################################################
if [ ! -s $WDIR/t000_.msa0.a3m ]
then
echo "Running HHblits"
$PIPEDIR/input_prep/make_msa.sh $IN $WDIR $CPU $MEM > $WDIR/log/make_msa.stdout 2> $WDIR/log/make_msa.stderr
fi
############################################################
# 2. predict secondary structure for HHsearch run
############################################################
if [ ! -s $WDIR/t000_.ss2 ]
then
echo "Running PSIPRED"
$PIPEDIR/input_prep/make_ss.sh $WDIR/t000_.msa0.a3m $WDIR/t000_.ss2 > $WDIR/log/make_ss.stdout 2> $WDIR/log/make_ss.stderr
fi
############################################################
# 3. search for templates
############################################################
DB=/NewWorkSpace/rosettafold/pdb100_2021Mar03/pdb100_2021Mar03
if [ ! -s $WDIR/t000_.hhr ]
then
echo "Running hhsearch"
HH="hhsearch -b 50 -B 500 -z 50 -Z 500 -mact 0.05 -cpu $CPU -maxmem $MEM -aliw 100000 -e 100 -p 5.0 -d $DB"
cat $WDIR/t000_.ss2 $WDIR/t000_.msa0.a3m > $WDIR/t000_.msa0.ss2.a3m
$HH -i $WDIR/t000_.msa0.ss2.a3m -o $WDIR/t000_.hhr -atab $WDIR/t000_.atab -v 0 > $WDIR/log/hhsearch.stdout 2> $WDIR/log/hhsearch.stderr
fi
############################################################
# 4. end-to-end prediction
############################################################
if [ ! -s $WDIR/t000_.3track.npz ]
then
echo "Running end-to-end prediction"
python $PIPEDIR/network/predict_e2e.py \
-m $PIPEDIR/weights \
-i $WDIR/t000_.msa0.a3m \
-o $WDIR/t000_.e2e \
--hhr $WDIR/t000_.hhr \
--atab $WDIR/t000_.atab \
--input_t1d $WDIR/input_t1d \
--input_t2d $WDIR/input_t2d \
--count_1d $WDIR/count_1d \
--count_2d $WDIR/count_2d \
--t1d $WDIR/t1d \
--t2d $WDIR/t2d \
--db $DB 1> $WDIR/log/network.stdout 2> $WDIR/log/network.stderr
fi
echo "Done"
(4)批量化运行并输出结果
#!/bin/sh
:<<!
author:chukai
time:20220422
content:批量化跑一批全长的HLA序列
!
echo "starting extracting..."
# 1、input path:
path=/home/xxx/RoseTTAFold-main/FullLengthMHC/
# 2、output path
output_path=/home/xxx/RoseTTAFold-main/outputs/
for file in `ls /home/xxx/RoseTTAFold-main/FullLengthMHC`;do
# input sequence realpath
export name="${path}$file"
echo $file
# output path+output filename
export outname="${output_path}$file"
# 一体化脚本:具体请查看run_e2e_ver.sh中的代码说明
starttime=`date +'%Y-%m-%d %H:%M:%S'`
/home/chuk/RoseTTAFold-main/run_e2e_ver.sh ${name} ${outname}
endtime=`date +'%Y-%m-%d %H:%M:%S'`
start_seconds=$(date --date="$starttime" +%s);
end_seconds=$(date --date="$endtime" +%s);
echo "runtime: "$((end_seconds-start_seconds))"s"
done
echo "---------------------------------------------------------------------------------------------------------------------------------------------------------"运行过程中遇到的问题:
1、会占用很大的内存,可以在脚本中及时清理
参考博客:
服务器清理内存shell脚本![]()

#! /bin/bash
#说明
#echo 1 > /proc/sys/vm/drop_caches:表示清除pagecache,当前产链服务器缓存主要在这里。
#echo 2 > /proc/sys/vm/drop_caches:表示清除回收slab分配器中的对象(包括目录项缓存和inode缓存)。slab分配器是内核中管理内存的一种机制,其中很多缓存数据实现都是用的pagecache。
#echo 3 > /proc/sys/vm/drop_caches:表示清除pagecache和slab分配器中的缓存对象。
used=`free -m | awk 'NR==2' | awk '{print $3}'`
free=`free -m | awk 'NR==2' | awk '{print $4}'`
echo "===========================" >> /root/mem.log
date >> /root/mem.log
echo "Memory usage before | [Use:${used}MB][Free:${free}MB]" >> /root/mem.log
if [ $free -le 10000 ] ; then
sync && echo 1 > /proc/sys/vm/drop_caches
sync && echo 2 > /proc/sys/vm/drop_caches
sync && echo 3 > /proc/sys/vm/drop_caches
used_ok=`free -m | awk 'NR==2' | awk '{print $3}'`
free_ok=`free -m | awk 'NR==2' | awk '{print $4}'`
echo "Memory usage after | [Use:${used_ok}MB][Free:${free_ok}MB]" >> /root/mem.log
echo "OK" >> /root/mem.log
else
echo "Not required" >> /root/mem.log
fi
exit 1
2、显存不足,可以采用并行或者多显卡
# 单显卡
device = torch.device("cuda:0")
# 并行
model = nn.DataParallel(model)
# 多显卡
os.environ["CUDA_VISIBLE_DEVICES"] = "1,2,3,4"
model = Model(input_size, output_size)
if torch.cuda.device_count() > 1:
# 如果不用os.environ的话,GPU的可见数量仍然是包括所有GPU的数量
# 但是使用的还是只用指定的device_ids的GPU
print("Let's use", torch.cuda.device_count(), "GPUs!")
# dim = 0 [30, xxx] -> [10, ...], [10, ...], [10, ...] on 3 GPUs
model = nn.DataParallel(model, device_ids=[1, 2, 3])
参考博客:如何使用pytorch进行多GPU训练_林子要加油的博客-CSDN博客_pytorch多gpu训练[0]
3、Linux中清理显存占用
Linux显存占用无进程清理方法(附批量清理命令)[0]
sudo fuser -v /dev/nvidia* |awk '{for(i=1;i<=NF;i++)print "kill -9 " $i;}' | sudo sh输出文件如下:


5、AlphaFold2原理介绍和安装
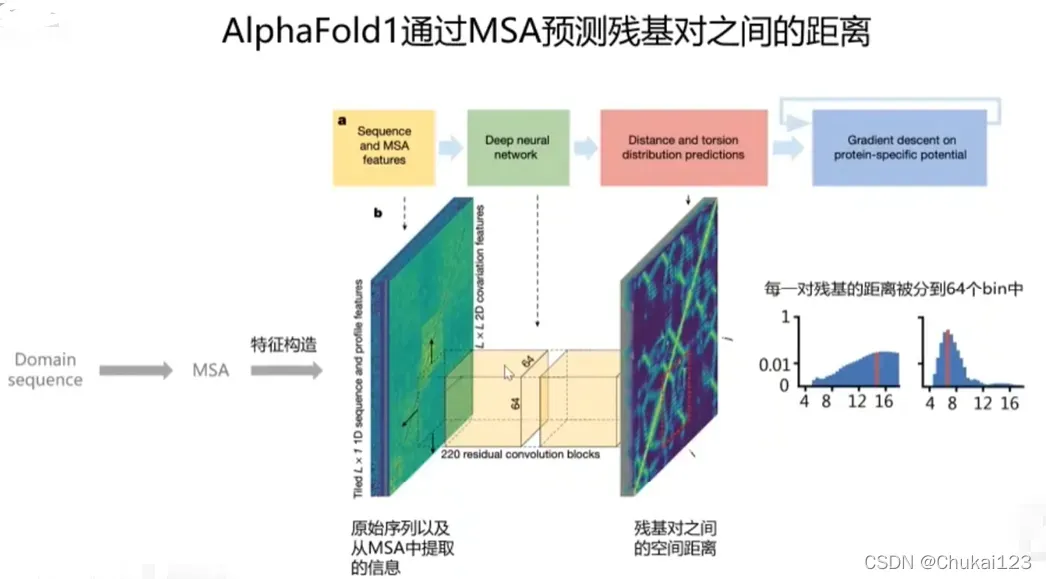

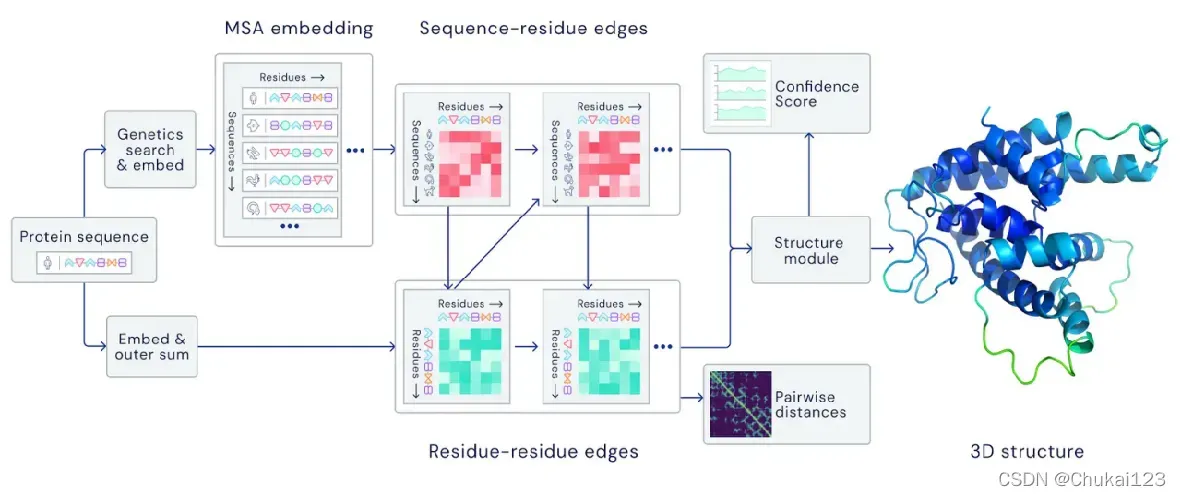

这一部分比较多,都整理到PPT中,可以下载查看!
AlphaFold2的原理和简单的安装过程[0]
文章出处登录后可见!