2017年5月,最开始是为了证明Scanpy可以复制Seurat的大部分聚类功能。数据3k PBMC来自健康的志愿者,可从10x Genomics免费获得。在unix系统上,可以取消注释并运行以下操作来下载和解压缩数据。最后一行创建一个用于保存已处理数据的目录write。
# !mkdir data
# !wget http://cf.10xgenomics.com/samples/cell-exp/1.1.0/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz -O data/pbmc3k_filtered_gene_bc_matrices.tar.gz
# !cd data; tar -xzf pbmc3k_filtered_gene_bc_matrices.tar.gz
# !mkdir write
导入相关工具,并做初始的设置:
import numpy as np
import pandas as pd
import scanpy as sc
sc.settings.verbosity = 3 # verbosity: errors (0), warnings (1), info (2), hints (3)
sc.logging.print_header()
sc.settings.set_figure_params(dpi=80, facecolor='white')
"""
scanpy==1.9.1 anndata==0.8.0 umap==0.5.3 numpy==1.21.6 scipy==1.8.1 pandas==1.4.2 scikit-learn==1.1.1 statsmodels==0.13.2 python-igraph==0.9.10 pynndescent==0.5.7
"""
声明h5ad用于存储分析结果:
results_file = 'write/pbmc3k.h5ad'
将计数矩阵(count matrix)读到Adata对象,adata包含数据及其注释信息(dataframe的obs和var等),它还具有基于HDF5的文件格式:h5ad。
adata = sc.read_10x_mtx(
'data/filtered_gene_bc_matrices/hg19/', # `.mtx`文件所在的目录
var_names='gene_symbols', # 用 gene 作为var
cache=True) # 开启缓存读写
"""
注意cache=Trure
... writing an h5ad cache file to speedup reading next time
下次读取就不会从count matrix读, 会直接从cache目录下的h5ad文件读(更快)
"""
注意,如果在函数sc.read_10x_mtx中指定参数var_names='gene_ids'时,下一个操作将是不必要的:
# 消除重复的列
adata.var_names_make_unique()
adata
"""
AnnData object with n_obs × n_vars = 2700 × 32738
var: 'gene_ids'
"""
对初始Adata的预处理
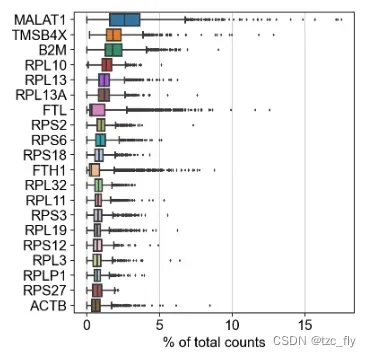
下图计算每一个基因在所有细胞中的平均表达量,并绘制了平均表达量前20的基因箱型图。
scanpy.pl.highest_expr_genes(adata, n_top=30)
计算每一个基因在所有细胞中的平均表达量。所有细胞中平均分数最高n_top的基因被绘制为箱形图。
箱型图是对每个基因在所有细胞中表达量分布的更详细描述。
sc.pl.highest_expr_genes(adata, n_top=20, )
然后进行基本的过滤(质量控制),使用两个工具:
sc.pp.filter_cells进行细胞的过滤,该函数保留至少有min_genes个基因(某个基因表达非0可判断存在该基因)的细胞,或者保留至多有max_genes个基因的细胞;sc.pp.filter_genes进行基因的过滤,该函数用于保留在至少min_cells个细胞中出现的基因,或者保留在至多max_cells个细胞中出现的基因;
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
adata
"""
AnnData object with n_obs × n_vars = 2700 × 13714
obs: 'n_genes'
var: 'gene_ids', 'n_cells'
"""
下一步是过滤线粒体核糖体基因(质量控制的选做步骤):这是一个很难把握的工作,需要结合自己项目的情况来做。不过通常有以下策略:
- 粗暴去除所有线粒体核糖体基因,直接去除包含”MT-”开头的基因。
- 选择阈值去除高表达量的细胞,阈值很大程度上取决于对自己项目的了解程度,因为不同器官组织提取的单细胞,线粒体基因平均水平不一样。
使用pp.calculate_qc_metrics,我们可以高效计算很多度量指标:
# 将线粒体基因组保存为注释 var.mt
adata.var['mt'] = adata.var_names.str.startswith('MT-')
adata.var['mt']
"""
AL627309.1 False
...
SRSF10-1 False
Name: mt, Length: 13714, dtype: bool
"""
# 计算指标, qc的var选择 var.mt
sc.pp.calculate_qc_metrics(adata, qc_vars=['mt'], percent_top=None, log1p=False, inplace=True)
adata
"""
注释中多了很多QC计算得到的信息
AnnData object with n_obs × n_vars = 2700 × 13714
obs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt'
var: 'gene_ids', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts'
"""
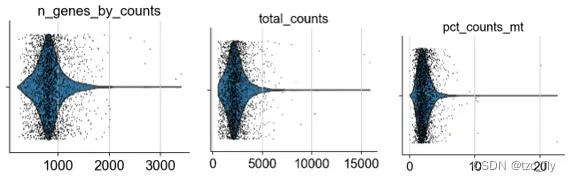
使用violinplot度量以下质量:
n_genes_by_counts:每个细胞中,有表达的基因的个数;total_counts:每个细胞的基因总计数(总表达量);pct_counts_mt:每个细胞中,线粒体基因表达量占该细胞所有基因表达量的百分比;
sc.pl.violin(adata, ['n_genes_by_counts', 'total_counts', 'pct_counts_mt'],
jitter=0.4, multi_panel=True)
下一步,我们移除线粒体基因表达过多(线粒体表达过多通常质量不好)或有表达的基因的个数过多(通常是测量成多细胞导致的,需要去除这种行数据)的细胞,先用散点图可视化这两个注释:
sc.pl.scatter(adata, x='total_counts', y='pct_counts_mt')
sc.pl.scatter(adata, x='total_counts', y='n_genes_by_counts')
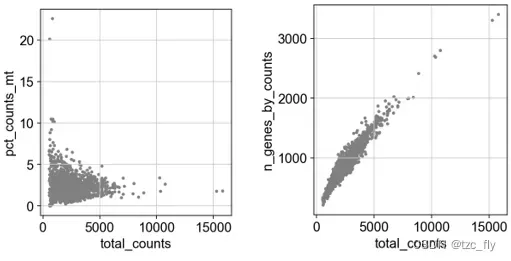
通过观察上图,我们对Adata切片:
adata = adata[adata.obs.n_genes_by_counts < 2500, :]
adata = adata[adata.obs.pct_counts_mt < 5, :]
adata
"""
View of AnnData object with n_obs × n_vars = 2638 × 13714
obs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt'
var: 'gene_ids', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts'
"""
下面我们对数据adata.X标准化,以便细胞之间的基因表达量具有可比性:
sc.pp.normalize_total(adata, target_sum=1e4)
函数normalize_total可以对每个细胞进行标准化,以便每个细胞在标准化后沿着基因方向求和具有相同的总数target_sum:
adata.X
array([[ 3., 3., 3., 6., 6.],
[ 1., 1., 1., 2., 2.],
[ 1., 22., 1., 2., 2.]], dtype=float32)
# 设置 target_sum=1 标准化后
X_norm
array([[0.14, 0.14, 0.14, 0.29, 0.29],
[0.14, 0.14, 0.14, 0.29, 0.29],
[0.04, 0.79, 0.04, 0.07, 0.07]], dtype=float32)
为了适当扩大表达的差异,我们对数据取对数:
sc.pp.log1p(adata)
下一步识别高变基因:
- 高变异基因就是highly variable features(HVGs),就是在细胞与细胞间进行比较,选择表达量差别最大的基因,Seurat使用FindVariableFeatures函数鉴定高变基因,这些基因在不同细胞之间的表达量差异很大(在一些细胞中高表达,在另一些细胞中低表达)。默认情况下,会返回2,000个高变基因用于下游的分析。
- 利用FindVariableFeatures函数,会计算一个mean-variance结果,也就是给出表达量均值和方差的关系并且得到top variable features,这一步的目的是鉴定出细胞与细胞之间表达量相差很大的基因,用于后续鉴定细胞类型。
- 标记基因 (marker gene),是一种已知功能或已知序列的基因,能够起着特异性标记的作用。
- 标记基因通常是高变基因中很小的子集;
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
"""
extracting highly variable genes
finished (0:00:00)
--> added
'highly_variable', boolean vector (adata.var) 注意该说明
'means', float vector (adata.var)
'dispersions', float vector (adata.var)
'dispersions_norm', float vector (adata.var)
"""
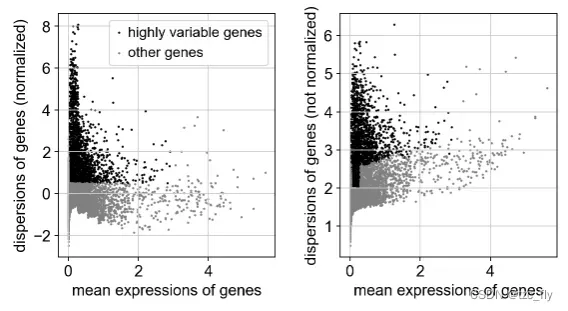
对高变基因可视化:
sc.pl.highly_variable_genes(adata)
在进一步筛选数据前,我们将当前adata保存到adata的元素raw下:
adata
"""
AnnData object with n_obs × n_vars = 2638 × 13714
obs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt'
var: 'gene_ids', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm'
uns: 'log1p', 'hvg'
"""
adata.raw = adata
然后我们对当前adata切片(基于bool型adata.var.highly_variable):
adata = adata[:, adata.var.highly_variable]
adata
"""
View of AnnData object with n_obs × n_vars = 2638 × 1838 注意列数变化
obs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt'
var: 'gene_ids', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm'
uns: 'log1p', 'hvg'
"""
我们对两个注释进行回归,即:
total_counts:每个细胞的基因总计数(总表达量);pct_counts_mt:每个细胞中,线粒体基因表达量占该细胞所有基因表达量的百分比;
# scipy最好是1.4.1
sc.pp.regress_out(adata, ['total_counts', 'pct_counts_mt'])
回归操作不会增加新的注释信息,我们只是为了演示,下一步我们按零均值单位方差标准化数据,并剪裁值超过标准差10的细胞。
sc.pp.scale(adata, max_value=10)
adata
"""
AnnData object with n_obs × n_vars = 2638 × 1838
obs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt'
var: 'gene_ids', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std'
uns: 'log1p', 'hvg'
"""
主成分分析
通过运行主成分分析(PCA)来降低数据的维数,这可以揭示主成分并对数据进行去噪:
sc.tl.pca(adata, svd_solver='arpack')
"""
computing PCA
on highly variable genes
with n_comps=50
finished (0:00:01)
"""
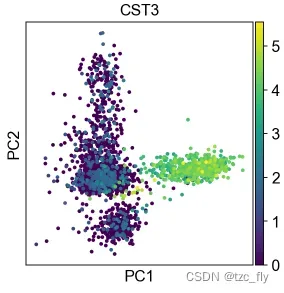
基于降维的数据,我们用散点图可视化,我们选取前两个主成分,并可视化基因CST3的表达量分布:
sc.pl.pca(adata, color='CST3')
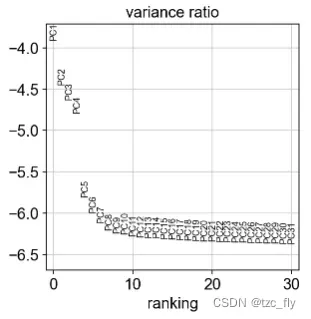
让我们检查单个PC(主成分)对数据总方差的贡献。这为我们提供了计算细胞的neighborhood relations时应考虑多少个PC的信息,例如在聚类函数sc.tl.louvain()或TSNEsc.tl.tsne()中使用的PC(主成分作为输入特征)。根据我们的经验,通常粗略估计PC的数量就可以了。
sc.pl.pca_variance_ratio(adata, log=True)
将结果保存到之前声明的文件results_file下,注意确保已经有目录write被创建好:
adata.write(results_file)
此时adata多了一个obsm注释X_pca:
adata
"""
AnnData object with n_obs × n_vars = 2638 × 1838
obs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt'
var: 'gene_ids', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std'
uns: 'log1p', 'hvg', 'pca'
obsm: 'X_pca'
varm: 'PCs'
"""
计算neighborhood graph
我们使用数据矩阵的PCA表示来计算细胞的邻域图。可以在这里简单地使用默认值。
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)
"""
computing neighbors
using 'X_pca' with n_pcs = 40
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:05)
"""
adata
"""
AnnData object with n_obs × n_vars = 2638 × 1838
obs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt'
var: 'gene_ids', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std'
uns: 'log1p', 'hvg', 'pca', 'neighbors'
obsm: 'X_pca'
varm: 'PCs'
obsp: 'distances', 'connectivities'
"""
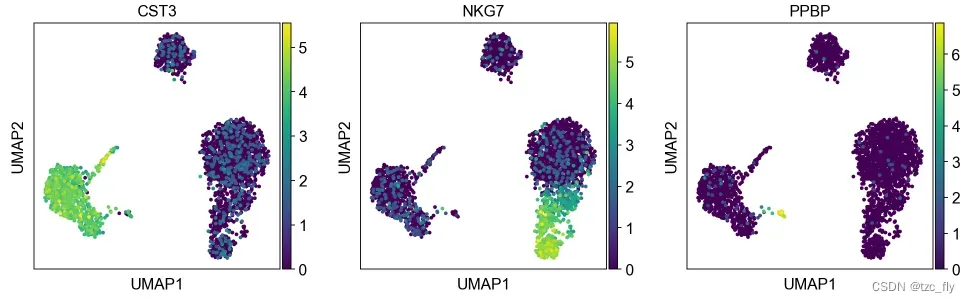
对neighborhood graph进行embedding
我们可以对graph进行embedding,比如使用UMAP进行降维,同时使用2维数据可以做可视化工作。UMAP比tSNE更符合流形的全局连通性,即更好地保持轨迹。
sc.tl.umap(adata)
"""
computing UMAP
finished: added
'X_umap', UMAP coordinates (adata.obsm) (0:00:03)
"""
sc.pl.umap(adata, color=['CST3', 'NKG7', 'PPBP'])
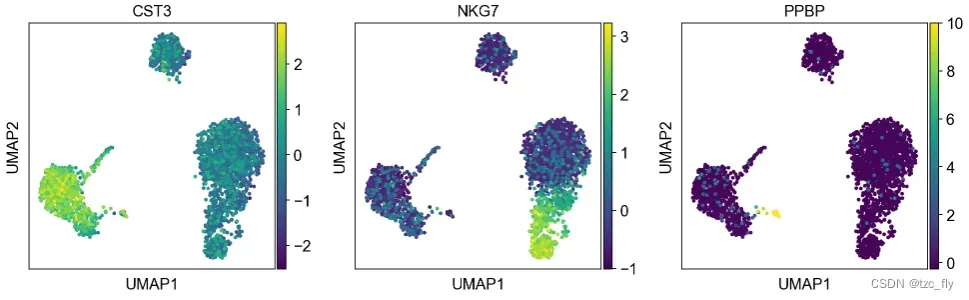
由于我们之前设置过adata.raw,之前的可视化是raw数据的。我们可以在可视化的时候设置”不使用raw数据”:
sc.pl.umap(adata, color=['CST3', 'NKG7', 'PPBP'], use_raw=False)
对neighborhood graph进行聚类
scanpy推荐Leiden图聚类方法。注意,Leiden聚类直接对细胞的neighborhood graph进行聚类。聚类结果显示9个簇:
sc.tl.leiden(adata)
"""
running Leiden clustering
finished: found 9 clusters and added
'leiden', the cluster labels (adata.obs, categorical) (0:00:00)
"""
然后我们用umap对leiden的聚类结果可视化:
sc.pl.umap(adata, color=['leiden', 'CST3', 'NKG7'])
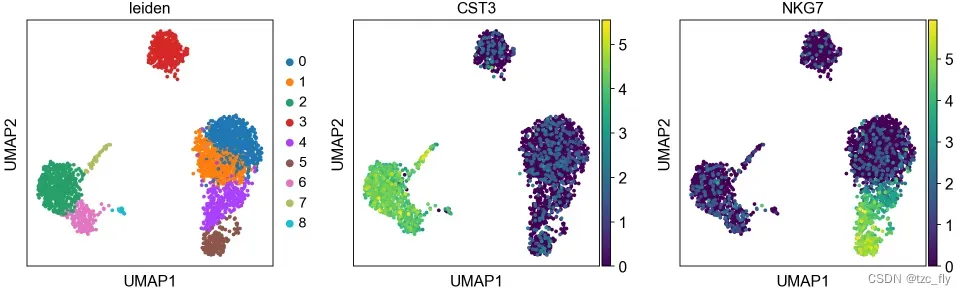
我们可以保存当前的adata:
adata.write(results_file)
找到marker基因(簇间比较)
现在我们计算每个簇中存在较高程度差异的基因的排名。默认情况下,使用adata.raw进行计算,在计算差异时,最简单最快的方式是t-检验:
sc.tl.rank_genes_groups(adata, 'leiden', method='t-test')
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
"""
ranking genes
finished: added to `.uns['rank_genes_groups']`
'names', sorted np.recarray to be indexed by group ids
'scores', sorted np.recarray to be indexed by group ids
'logfoldchanges', sorted np.recarray to be indexed by group ids
'pvals', sorted np.recarray to be indexed by group ids
'pvals_adj', sorted np.recarray to be indexed by group ids (0:00:00)
"""
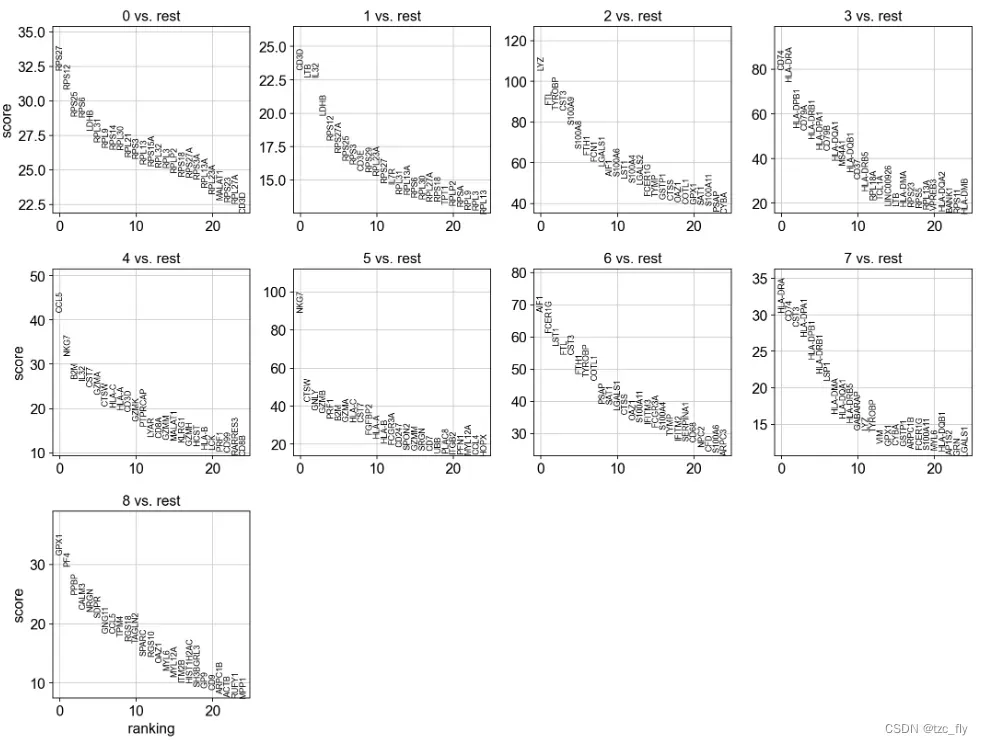
除了t-test(t检验),还有更多的检测方式,比如:wilcoxon。
sc.tl.rank_genes_groups(adata, 'leiden', method='wilcoxon')
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
保存当前adata:
adata.write(results_file)
另一种方式是用逻辑回归对基因进行rank,这与其他rank方法相比,逻辑回归使用了多变量,而传统方法是基于单变量的。
sc.tl.rank_genes_groups(adata, 'leiden', method='logreg')
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
差异分析是为了得到差异基因,他们都是潜在的marker基因。
我们收集各个研究文献中的结论,得到标记基因与典型细胞类型的关系:
| Markers | Cell |
|---|---|
| IL7R | CD4 T cells |
| CD14, LYZ | CD14+ Monocytes |
| MS4A1 | B cells |
| CD8A | CD8 T cells |
| GNLY, NKG7 | NK cells |
| FCGR3A, MS4A7 | FCGR3A+ Monocytes |
| FCER1A, CST3 | Dendritic Cells |
| PPBP | Megakaryocytes |
现在我们定义一个标记基因列表,以供以后参考。
marker_genes = ['IL7R', 'CD79A', 'MS4A1', 'CD8A', 'CD8B', 'LYZ', 'CD14',
'LGALS3', 'S100A8', 'GNLY', 'NKG7', 'KLRB1',
'FCGR3A', 'MS4A7', 'FCER1A', 'CST3', 'PPBP']
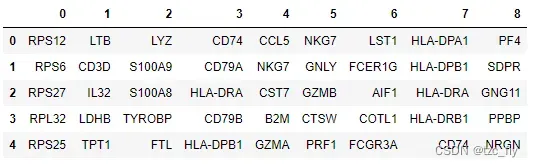
重新加载由 Wilcoxon rank 的测试结果一起保存的对象。
adata = sc.read(results_file)
然后用表格形式显示每个簇中,排名前5的差异基因:
pd.DataFrame(adata.uns['rank_genes_groups']['names']).head(5)
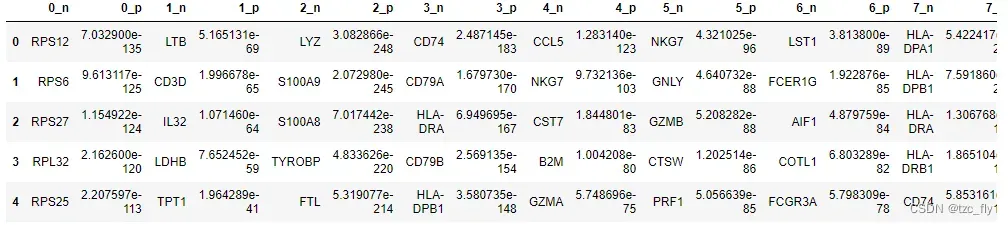
更详细的,我们也可以显示差异基因的名称和差异度:
result = adata.uns['rank_genes_groups']
groups = result['names'].dtype.names
pd.DataFrame(
{group + '_' + key[:1]: result[key][group]
for group in groups for key in ['names', 'pvals']}).head(5)
我们可以用小提琴图显示更细节的差异基因分布,下面我们为每个簇,展示前20个差异基因在该簇和剩余簇的分布差异:
from matplotlib.pyplot import rc_context
with rc_context({'figure.figsize': (9, 1.5)}):
sc.pl.rank_genes_groups_violin(adata, n_genes=20, jitter=False)
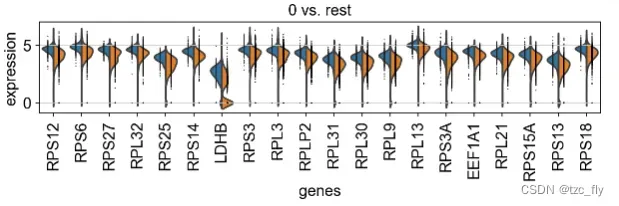
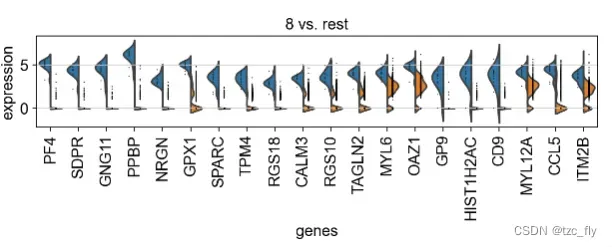
由于篇幅有限,我们仅在本篇内容里展示(簇0和剩余)和(簇8和剩余)。
如果想要直观地显示某个基因在不同簇中的表达,可以使用:
sc.pl.violin(adata, ['CST3', 'NKG7', 'PPBP'], groupby='leiden')
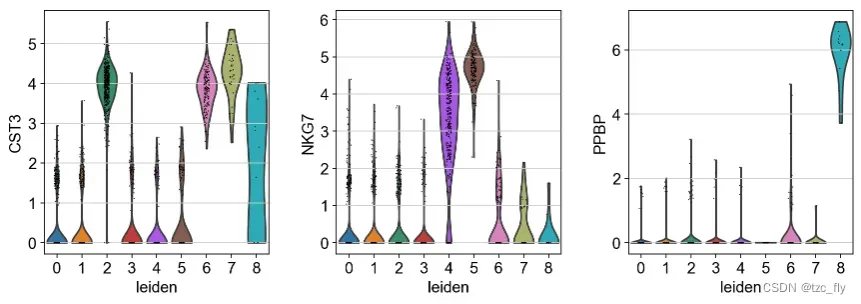
我们为每个簇选择top差异基因,与”marker基因-细胞”资料库对比得到细胞类型,然后,我们输入簇名称列表(手动将名称与序号顺序对应),再次可视化。
首先,将名称覆盖到adata的聚类结果标签字段"leiden",我在本次实验中只找到3个类型,但是其实我们可以用不同差异计算方式得到差异基因,然后再对比得到细胞类型,直到找满为止:
new_cluster_names = [
'0', '1', '2: CD14+Monocytes',
'3', '4',
'5: NK', '6',
'7', '8: Megakaryocytes']
adata.rename_categories('leiden', new_cluster_names)
再次可视化这些簇:
sc.pl.umap(adata, color='leiden', legend_loc='on data', title='', frameon=False)
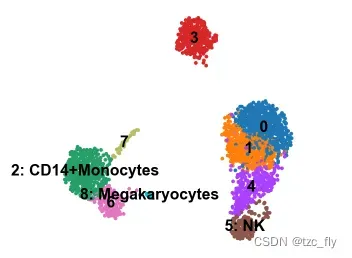
假设我们已经分配好了所有细胞类型,下一步我们用tracksplot验证基因与细胞的关系:
sc.pl.rank_genes_groups_tracksplot(adata, n_genes=3)
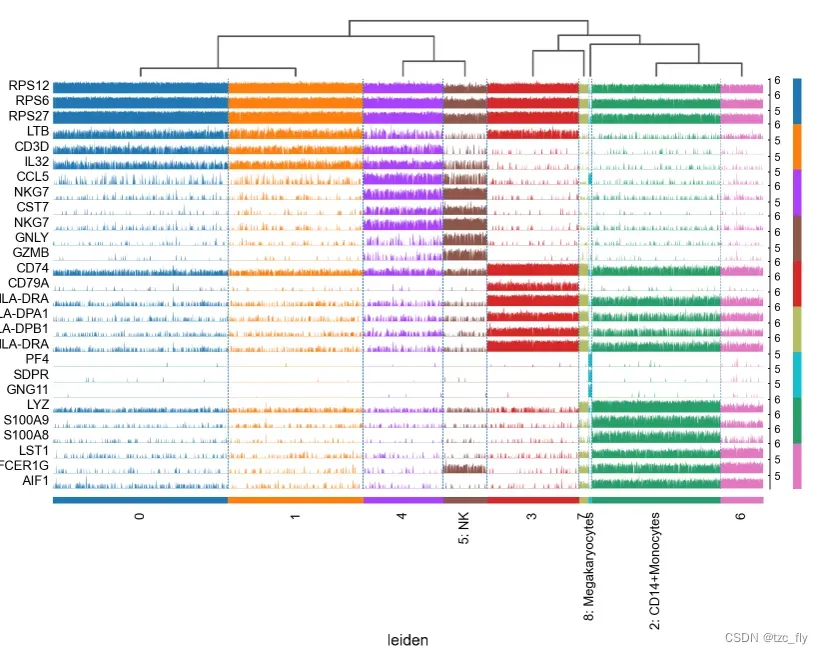
tracksplot直观显示了基因与细胞的分布关系,可以进一步帮我们研究标签分布的修正工作。比如簇0和簇1分布很相似,其实它们应该是同一类细胞。
文章出处登录后可见!