下面的tutorial描述了一种基于PCA的简单方法用于集成数据(integrating data),我们称之为ingest,并将其与BBKNN进行了比较。此外,BBKNN可以与Scanpy工作流良好结合,可通过bbknn接口访问。
ingest函数假设有一个带注释的参考数据集,该数据集捕获了感兴趣的生物变异性。ingest是在参考数据上拟合模型,并使用它来投影新数据。目前,该模型是一个结合了邻居查找搜索树(neighbor lookup search tree)的PCA或者UMAP。ingest有以下特点:
- 由于ingest操作简单,过程清晰,因此工作流透明且快速。
- 与BBKNN相似,ingest使数据矩阵本身保持不变。因为ingest是在embedding空间做分析;
- 与BBKNN不同,ingest在解决了标签映射的同时,维持了可能具有特定簇或轨迹等所需属性的embedding(因为BBKNN是简单粗暴的对齐embedding)。
我们将这种非对称数据集integration称为ingesting,将注释从带注释的reference adata_ref 提取到仍然缺少此注释的adata中。它不同于学习以对称方式集成数据集的联合表示,如BBKNN、Scanorma、Conos、CCA或 conditional VAE。
BBKNN
其中,关于BBKNN来自论文:“BBKNN: fast batch alignment of single cell transcriptomes”;
scRNA序列分析中的一个常见步骤是识别邻域图(neighbourhood graph),通常是在主成分空间中识别每个细胞的k个最近邻。该图很好地近似了细胞群体结构,为不同的下游分析提供了基础。这包括聚类、降维可视化和伪时间轨迹推断。
然而,批次效应增加的实验变化通常会导致细胞无法跨批次连接到相同的细胞类型,从而导致该图结构的扭曲和破裂。这会导致上述所有下游分析选项出现严重问题。
BBKNN修改了邻域构建步骤,以生成一个跨所有批次数据平衡的图。这种方法将邻居网络视为数据的主要表示形式。对于每个细胞,BBKNN图是通过独立地为每个设备提供的批次(单个数据集)中的每个细胞找到k个最近邻居来构建的,这使得每个细胞在每个批次中都有一个独立的邻居池。同类细胞的这些邻居都被加入列表联系起来。
注意:BBKNN会将列表中的元素用简单快速的方式对齐,即对齐不同批次的同类细胞。所以才说BBKNN实现了数据整合。
BBKNN的主要假设是,批次间至少存在一些相同类型的细胞,并且批次效应引起的批次间相同细胞类型之间的差异小于批次内不同类型细胞之间的差异。在这种情况下,图的构造将跨批次将相似的细胞类型分组在一起,同时将不相关的细胞类型分开。
导入初始信息:
import scanpy as sc
import pandas as pd
import seaborn as sns
sc.settings.verbosity = 1 # verbosity: errors (0), warnings (1), info (2), hints (3)
sc.logging.print_versions()
sc.settings.set_figure_params(dpi=80, frameon=False, figsize=(3, 3), facecolor='white')
PBMCs
我们考虑一个带注释的 referenced dataset adata_ref 和一个要查询label和embedding的数据集 adata。通常,adata_ref的大小是大于adata的,这才能确保adata_ref包含足够的细胞(即足够的生物多样性)。
# PBMCs 3k(已经处理)
adata_ref = sc.datasets.pbmc3k_processed()
# 700个细胞和765个高变基因, 此数据集已经过预处理和UMAP计算
adata = sc.datasets.pbmc68k_reduced()
adata_ref
"""
AnnData object with n_obs × n_vars = 2638 × 1838
obs: 'n_genes', 'percent_mito', 'n_counts', 'louvain'
var: 'n_cells'
uns: 'draw_graph', 'louvain', 'louvain_colors', 'neighbors', 'pca', 'rank_genes_groups'
obsm: 'X_pca', 'X_tsne', 'X_umap', 'X_draw_graph_fr'
varm: 'PCs'
obsp: 'distances', 'connectivities'
"""
为了能应用sc.tl.ingest,数据集需要具有相同的var,我们需要计算交集:
# 求交集
var_names = adata_ref.var_names.intersection(adata.var_names)
var_names
"""
Index(['TNFRSF4', 'SRM', 'TNFRSF1B', 'EFHD2', 'C1QA', 'C1QB', 'STMN1',
'MARCKSL1', 'SMAP2', 'PRDX1',
...
'EIF3D', 'LGALS2', 'ADSL', 'TTC38', 'TYMP', 'ATP5O', 'TTC3', 'SUMO3',
'S100B', 'PRMT2'],
dtype='object', name='index', length=208)
"""
对数据切片:
adata_ref = adata_ref[:, var_names]
adata = adata[:, var_names]
adata
"""
View of AnnData object with n_obs × n_vars = 700 × 208
obs: 'bulk_labels', 'n_genes', 'percent_mito', 'n_counts', 'S_score', 'G2M_score', 'phase', 'louvain'
var: 'n_counts', 'means', 'dispersions', 'dispersions_norm', 'highly_variable'
uns: 'bulk_labels_colors', 'louvain', 'louvain_colors', 'neighbors', 'pca', 'rank_genes_groups'
obsm: 'X_pca', 'X_umap'
varm: 'PCs'
obsp: 'distances', 'connectivities'
"""
adata_ref
"""
View of AnnData object with n_obs × n_vars = 2638 × 208
obs: 'n_genes', 'percent_mito', 'n_counts', 'louvain'
var: 'n_cells'
uns: 'draw_graph', 'louvain', 'louvain_colors', 'neighbors', 'pca', 'rank_genes_groups'
obsm: 'X_pca', 'X_tsne', 'X_umap', 'X_draw_graph_fr'
varm: 'PCs'
obsp: 'distances', 'connectivities'
"""
根据reference data得到model和graph(这里是PCA、neighbors、UMAP)是ingest之前需要的计算:
sc.pp.pca(adata_ref)
sc.pp.neighbors(adata_ref)
sc.tl.umap(adata_ref)
adata_ref
"""
AnnData object with n_obs × n_vars = 2638 × 208
obs: 'n_genes', 'percent_mito', 'n_counts', 'louvain'
var: 'n_cells'
uns: 'draw_graph', 'louvain', 'louvain_colors', 'neighbors', 'pca', 'rank_genes_groups', 'umap'
obsm: 'X_pca', 'X_tsne', 'X_umap', 'X_draw_graph_fr'
varm: 'PCs'
obsp: 'distances', 'connectivities'
"""
用UMAP可视化流形:
sc.pl.umap(adata_ref, color='louvain')
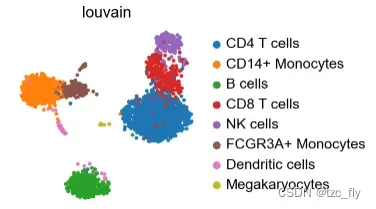
Mapping PBMCs using ingest
我们基于已选择的representation,将label和embedding从adata_ref映射到adata。在ingest中,使用adata_ref.obsm['X_pca']去映射簇标签和UMAP坐标(即X_umap)。
sc.tl.ingest(adata, adata_ref, obs='louvain')
用法:sc.tl.ingest(),用于从reference data映射labels和embeddings到new data(将new data映射到reference data的PCA空间)。把adata投影到已拟合在adata_ref上的PCA轴。该函数使用knn分类器映射adata的labels,使用UMAP或者PCA映射adata的embeddings。我们将这种非对称数据集集成称为将注释从参考数据ingest到新数据。
scanpy.tl.ingest(adata, adata_ref, obs=None, embedding_method=('umap', 'pca'), labeling_method='knn')
- 参数
embedding_method:使用adata_ref的umap或pca参数(即adata_ref.uns['pca']和adata_ref.uns['umap'])去映射得到adata的embedding。 - 参数
labeling_method:使用knn去映射得到adata的label(训练数据或参考数据为adata_ref的embedding,测试数据为映射后的adata的embedding,根据相似度赋予adata标签)。 - 参数
obs:映射使用的标签键,比如cell_type,louvain等(从adata_ref映射到adata中)。
在调用ingest前,需要对adatat_ref运行neighbors()。(neighbors()用于计算观测的neighborhood graph,这是ingest计算前的处理)
import scanpy as sc
sc.pp.neighbors(adata_ref)
sc.tl.umap(adata_ref)
sc.tl.ingest(adata, adata_ref, obs='cell_type')
然后对齐颜色:
# 将adata的聚类颜色与adata_ref的对应
adata.uns['louvain_colors'] = adata_ref.uns['louvain_colors']
adata.uns['louvain_colors']
"""
array(['#1f77b4', '#ff7f0e', '#2ca02c', '#d62728', '#9467bd', '#8c564b',
'#e377c2', '#bcbd22'], dtype='<U7')
"""
可视化ingest处理后的adata:
# UMAP可视化, louvain是ingest的obs, bulk_labels是GT
sc.pl.umap(adata, color=['louvain', 'bulk_labels'], wspace=0.5)
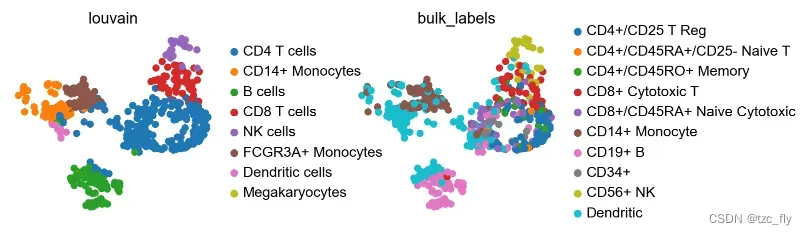
可以看到,ingest后的adata的类型来自adata_ref,与真实标签对比,其实数据的簇已经得到了有规则的区分映射,只有少数的比如Dendritic cell还是不明确的。
观察上图,bulk_labels其实是louvain的更细粒度划分。
最后开始整合两个数据集,即拼接矩阵:
adata_concat = adata_ref.concatenate(adata, batch_categories=['ref', 'new'])
将拼接后的矩阵固定类别的顺序和颜色,让其与adata_ref一致:
adata_concat.obs.louvain = adata_concat.obs.louvain.astype('category')
# 固定category顺序
adata_concat.obs.louvain.cat.reorder_categories(adata_ref.obs.louvain.cat.categories, inplace=True)
adata_ref.obs.louvain.cat.categories
"""
Index(['CD4 T cells', 'CD14+ Monocytes', 'B cells', 'CD8 T cells', 'NK cells',
'FCGR3A+ Monocytes', 'Dendritic cells', 'Megakaryocytes'],
dtype='object')
"""
# 固定category的颜色
adata_concat.uns['louvain_colors'] = adata_ref.uns['louvain_colors']
最后可视化聚合后的数据:
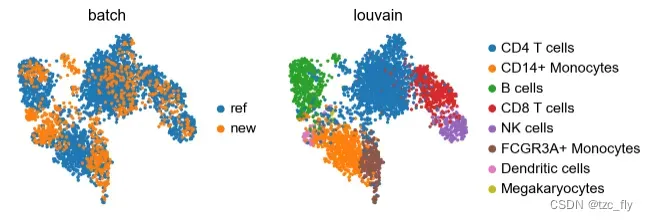
sc.pl.umap(adata_concat, color=['batch', 'louvain'])

虽然来自新数据(左图橙色)的树突状细胞(dendritic cell)簇中似乎存在一些批次效应(batch-effect),但新数据在其他方面的映射相对均匀。
- 树突状细胞,粉红色的散点不够聚集,在橙色附近和绿色附近都出现了;
- 其实看adata_ref,粉色点不聚集的现象其实已经存在;
批次效应
不同时间、不同操作者、不同试剂、不同仪器导致的实验误差,反映到细胞的表达量上就是批次效应,这个很难去除但可以缩小。
校正批次效应的目的就是:减少数据集之间的差异,尽量让多个数据集重新组合在一起,这样下游分析就可以只考虑生物学差异因素。
巨核细胞(megakaryoctes)仅存在于adata_ref中,adata中没有细胞映射到巨核细胞上。如果交换参考数据和查询数据,巨核细胞将不再能被检测到。这是一种极端情况,因为参考数据非常小导致;
我们应该始终注意,参考数据是否包含足够的生物多样性,以有意义地包含了查询数据。
Using BBKNN
BBKNN:Batch balanced KNN,BBKNN是一种快速直观的去除批次效应工具,可直接用于scanpy工作流。它是scanpy.pp.neighbors()的一种替代方法,这两个功能都是创建一个neighbors graph,以便后续在聚类、pseudo time计算和UMAP可视化中使用。标准方法首先确定整个数据集中每个细胞的k个最近邻,然后将候选细胞转换为指数相关连接,然后作为进一步分析的基础。如果数据中存在批次效应(可能是由于不同的数据采集技术、协议变更,甚至是特别严重的操作员影响),那么将很难在不同批次中链接相应的细胞类型。
注意这里的batch其实是一个数据集。
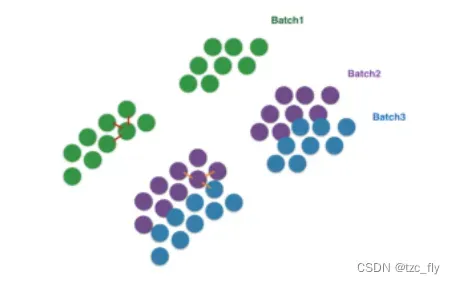
因此,BBKNN通过获取每个细胞并分别识别每个批次中的(较小的)k个最近邻居而不是在整个整合的数据集上分析。然后,每个批次的这些最近邻域将合并到细胞的邻居列表中。这有助于在不同批次的类似细胞之间建立联系,而无需改变矩阵的表达量或PCA空间。
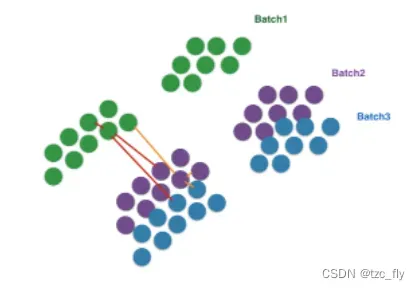
使用bbknn聚合数据:
sc.tl.pca(adata_concat)
%%time
sc.external.pp.bbknn(adata_concat, batch_key='batch')
"""
CPU times: total: 984 ms
Wall time: 979 ms
"""
sc.tl.umap(adata_concat)
sc.pl.umap(adata_concat, color=['batch', 'louvain'])
BBKNN也不能在new data中分配到巨核细胞(megakaryoctes)群。然而,BBKNN似乎能更均匀地混合ref和new数据的分布。
总结
基于ingest的单细胞数据整合就像是域适应任务,如果adata_ref包含的生物多样性是我们足够置信的,我们也许可以用域适应方法移动adata的分布到adata_ref上,再整合数据。
ingest是基于PCA的,将adata映射到adata_reference的PCA空间(基于adata_reference的PCA坐标系进行投影),然后对标签和各种embedding进行映射(即映射到和adata_reference空间一样的域下)。直观看,这种方法过于简单,我们或许应该在数据整合上沿用ingest的宏观策略,但是在ingest这一步骤上进行修改,改成当前流行的无监督域适应方法。
Pancreas
下面我们将使用另一个领域的数据实现数据整合工作,它包含来自4项不同研究(Segerstolpe16、Baron16、Wang16、Muraro16)的人类胰腺数据,这些数据已被用于单细胞数据集整合的开创性论文中,此后也在多次使用。
import scanpy as sc
import pandas as pd
import seaborn as sns
sc.settings.verbosity = 1 # verbosity: errors (0), warnings (1), info (2), hints (3)
sc.logging.print_versions()
sc.settings.set_figure_params(dpi=80, frameon=False, figsize=(3, 3), facecolor='white')
加载数据,没有下载会根据url自动下载:
# 此批次整合已在基因上相交
adata_all = sc.read('data/pancreas.h5ad', backup_url='https://www.dropbox.com/s/qj1jlm9w10wmt0u/pancreas.h5ad?dl=1')
adata_all.shape
"""
(14693, 2448)
"""
检查这些研究中观察到的细胞类型:
counts = adata_all.obs.celltype.value_counts()
counts
"""
alpha 4214
beta 3354
ductal 1804
acinar 1368
not applicable 1154
delta 917
gamma 571
endothelial 289
activated_stellate 284
dropped 178
quiescent_stellate 173
mesenchymal 80
macrophage 55
PSC 54
unclassified endocrine 41
co-expression 39
mast 32
epsilon 28
mesenchyme 27
schwann 13
t_cell 7
MHC class II 5
unclear 4
unclassified 2
Name: celltype, dtype: int64
"""
为了简化可视化,让我们删除5个少数类:
minority_classes = counts.index[-5:].tolist() # get the minority classes
adata_all = adata_all[ # actually subset
~adata_all.obs.celltype.isin(minority_classes)] # ~表示取反
adata_all.obs.celltype.cat.reorder_categories( # reorder according to abundance
counts.index[:-5].tolist(), inplace=True)
adata_all
"""
View of AnnData object with n_obs × n_vars = 14662 × 2448
obs: 'celltype', 'sample', 'n_genes', 'batch', 'n_counts', 'louvain'
var: 'n_cells-0', 'n_cells-1', 'n_cells-2', 'n_cells-3'
uns: 'celltype_colors', 'louvain', 'neighbors', 'pca', 'sample_colors'
obsm: 'X_pca', 'X_umap'
varm: 'PCs'
obsp: 'distances', 'connectivities'
"""
查看批次效应
注意我们的adata_all已经是4个批次的直接整合结果,即obs['batch']的值为:
index
human1_lib1.final_cell_0001-0 0
human1_lib1.final_cell_0002-0 0
human1_lib1.final_cell_0003-0 0
human1_lib1.final_cell_0004-0 0
human1_lib1.final_cell_0005-0 0
..
reads.29499-3 3
reads.29500-3 3
reads.29501-3 3
reads.29502-3 3
reads.29503-3 3
Name: batch, Length: 14662, dtype: category
Categories (4, object): ['0', '1', '2', '3']
我们用umap可视化这个直接整合过的数据:
sc.pp.pca(adata_all)
sc.pp.neighbors(adata_all)
sc.tl.umap(adata_all)
# palette用于选择颜色
sc.pl.umap(adata_all, color=['batch', 'celltype'], palette=sc.pl.palettes.vega_20_scanpy)
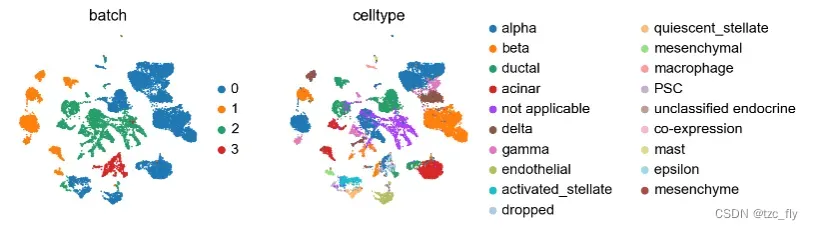
结合batch和celltype两个图,可以看到批次效应:比如alpha细胞,本是同一类,但却在celltype中呈现出多个距离较远的簇(这是由于它们存在于不同批次里,不同批次之间存在偏移)。
BBKNN
我们用BBKNN整合4个批次:
%%time
sc.external.pp.bbknn(adata_all, batch_key='batch')
"""
CPU times: total: 1.58 s
Wall time: 1.33 s
"""
sc.tl.umap(adata_all)
sc.pl.umap(adata_all, color=['batch', 'celltype'])
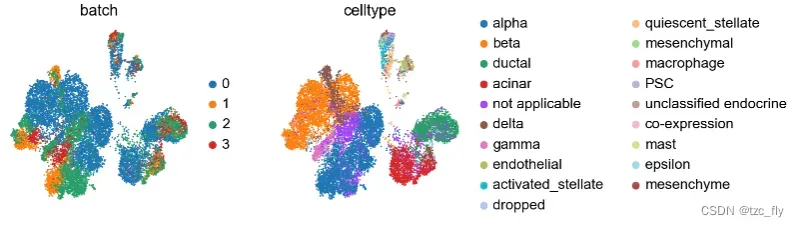
可以看到一定程度上,批次效应得到了削弱。
BBKNN依靠建立关联列表,并用简单方法快速对齐列表元素。可以看出BBKNN确实属于简便但较粗糙的整合方法。
如果希望从一个reference数据集开始以更迭代的方式执行整合,可以使用ingest。
使用ingest映射到reference批次
与之前一样,在参考批次上训练的模型将解释在其中观察到的生物变化。我们选择批次0作为参考批次:
adata_ref = adata_all[adata_all.obs.batch == '0']
计算PCA,邻居图(用于UMAP,UMAP方法回顾其他算法-UMAP),UMAP:
sc.pp.pca(adata_ref)
sc.pp.neighbors(adata_ref)
sc.tl.umap(adata_ref)
sc.pl.umap(adata_ref, color='celltype')
注意,我们的参考批次包含所有批次中19种细胞类型中的12种。

将参考数据中的标签(如“celltype”)和embedding(如“X_pca”和“X_umap”)映射到query批次上。
adatas = [adata_all[adata_all.obs.batch == i].copy() for i in ['1', '2', '3']]
sc.settings.verbosity = 2 # a bit more logging
for iadata, adata in enumerate(adatas):
print(f'... integrating batch {iadata+1}')
# celltype_orig保存批次原始的细胞类型
adata.obs['celltype_orig'] = adata.obs.celltype
sc.tl.ingest(adata, adata_ref, obs='celltype')
"""
... integrating batch 1
running ingest
finished (0:00:13)
... integrating batch 2
running ingest
finished (0:00:07)
... integrating batch 3
running ingest
finished (0:00:02)
"""
现在,每个query批次都带有已与adata_ref关联的注释。通过concat,我们可以一起查看整合的数据。
adata_concat = adata_ref.concatenate(adatas)
adata_concat.obs.celltype = adata_concat.obs.celltype.astype('category')
adata_concat.obs.celltype.cat.reorder_categories(adata_ref.obs.celltype.cat.categories, inplace=True) # fix category ordering
adata_concat.uns['celltype_colors'] = adata_ref.uns['celltype_colors'] # fix category coloring
sc.pl.umap(adata_concat, color=['batch', 'celltype'])
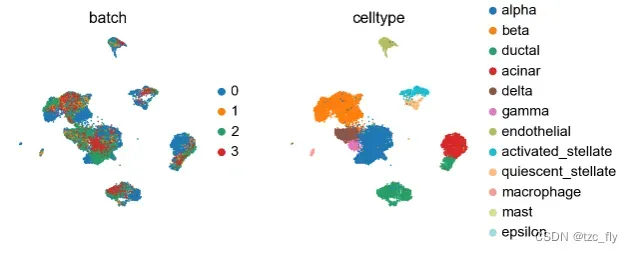
与BBKNN结果相比,这是以一种能更明显保持簇结构的方式。因为ingest采用了域适应的思想,而不是BBKNN的简单对齐两个域。
一致性评估
现在我们保留数据中的query批次。即去除adata_reference(第0批次):
adata_query = adata_concat[adata_concat.obs.batch.isin(['1', '2', '3'])]
下面的图有点难看出某些现象,因此,请转到本文下面的混淆矩阵。
sc.pl.umap(
adata_query, color=['batch', 'celltype', 'celltype_orig'], wspace=0.4)

让我们首先关注对于批次的原始细胞类型,其和reference data保持一致的那些细胞类型,以简化混淆矩阵的计算。
obs_query = adata_query.obs
conserved_categories = obs_query.celltype.cat.categories.intersection(obs_query.celltype_orig.cat.categories) # intersected categories
obs_query_conserved = obs_query.loc[obs_query.celltype.isin(conserved_categories) & obs_query.celltype_orig.isin(conserved_categories)] # intersect categories
obs_query_conserved.celltype.cat.remove_unused_categories(inplace=True) # remove unused categoriyes
obs_query_conserved.celltype_orig.cat.remove_unused_categories(inplace=True) # remove unused categoriyes
obs_query_conserved.celltype_orig.cat.reorder_categories(obs_query_conserved.celltype.cat.categories, inplace=True) # fix category ordering
pd.crosstab(obs_query_conserved.celltype, obs_query_conserved.celltype_orig)
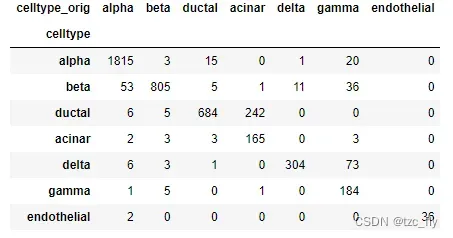
总的来说,细胞类型按预期映射。主要的例外是原始注释中的腺泡细胞(acinar cell)大部分被ingest认为是导管细胞(ductal cell)。然而,已经观察到参考数据的腺泡细胞和导管细胞就具有相似特征,这解释了这种差异,并表明参考注释中就存在潜在的不一致性。
可视化批次间的分布
对于批次的分布比较,Scanpy提供了方便的可视化:
- 密度图,density plot;
- 各个批次的直接可视化;
首先,我们可视化密度图:
sc.tl.embedding_density(adata_concat, groupby='batch')
"""
computing density on 'umap'
"""
sc.pl.embedding_density(adata_concat, groupby='batch')
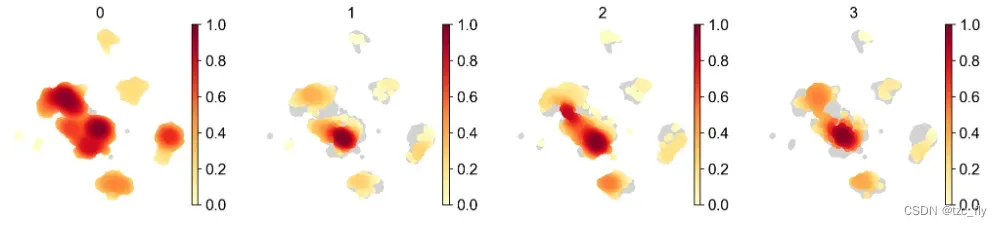
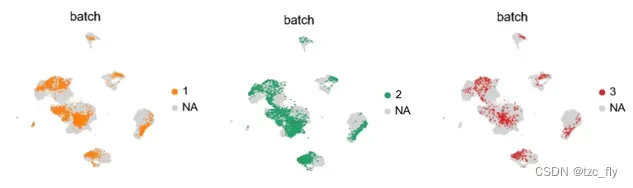
另外,我们可以直接可视化各个批次在所有数据中的分布:
for batch in ['1', '2', '3']:
sc.pl.umap(adata_concat, color='batch', groups=[batch])
文章出处登录后可见!