之前发过一个一对多的批量对接批处理程序(点我看一个受体对多个配体的程序),有小伙伴提出要做个多对多的程序,而且多对多也更常用一些。本文就此需求实现一下,同时还整合了数据结果的可视化,用热图的形式将多对多对接结果呈现出来。
另外,如果不想安装python,也可用批处理在批量对接完成后生成excel可读的文件,进行后续的排序和筛选(点我看)。
多对多批量对接
一、新建一个文件夹
新建一个文件夹,将我们整理好的配体和受体文件分别放在receptor和ligand两个文件夹内。将vina相关程序也放进来。
受体对接口袋的信息包括了xyz坐标等,要自己找到并写入到一个txt文件中,对接口袋信息举例如下
center_x = 187.9
center_y = -10.8
center_z = 61.7
size_x = 24.4
size_y = 23.6
size_z = 37.3
num_modes = 9
需要注意的是将受体的对接口袋信息也放入receptor文件中,并与pdbqt文件的文件名一致。
二、建立批处理程序
回到新建的文件夹中,新建一个txt文件,将以下代码复制进去
@echo
for %%r in (receptor\*.pdbqt) do (
for %%l in (ligand\*.pdbqt) do (
if not exist results mkdir results
vina --config receptor\%%~nr.txt --receptor %%r --ligand %%l --out results\%%~nr_2_%%~nl.pdbqt --log results\%%~nr_2_%%~nl.txt
timeout 10))
exit
如果不需要电脑休息,可将
timeout 10删去,最外面的括号一定注意不要删掉。
保存后,任意给他一个名字dock.txt也很,其他也好,这不重要。重要的是要把后缀名.txt改为.bat。有的人不喜欢显示后缀名,需要进入到文件夹选项中,把后缀名显示以便更改。
三、双击运行
然后,双击这个.bat,你的电脑就开始起飞了。
运行结束后,结果会保存在一个叫results的文件夹中。如图所示。
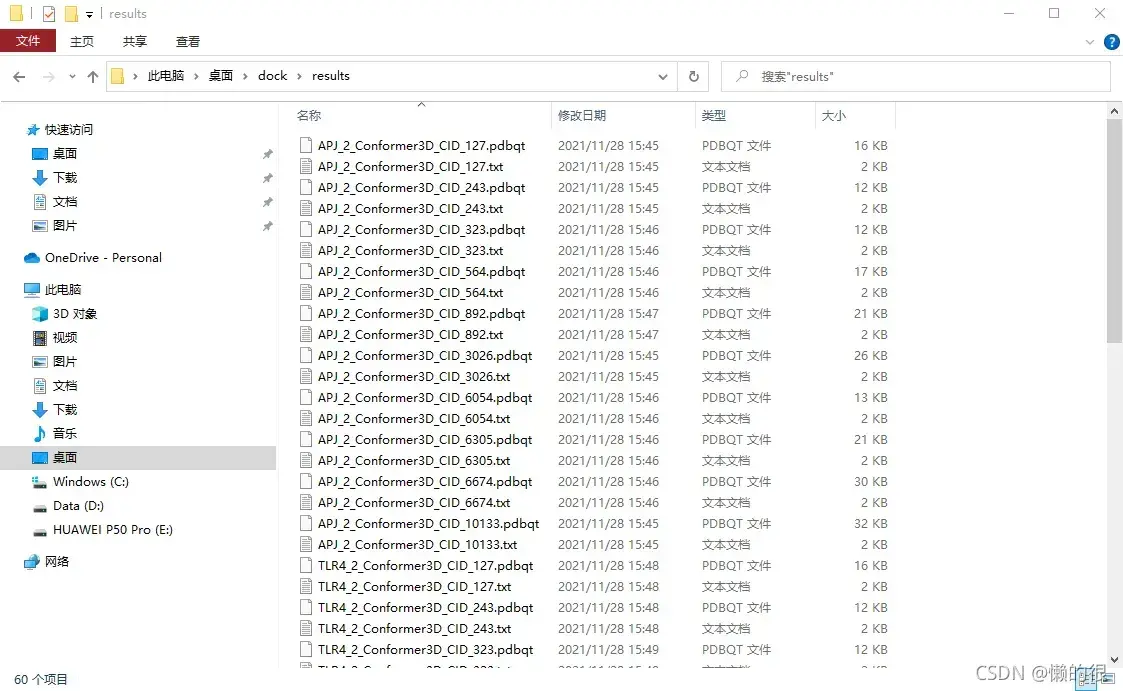
结果的文件名命名规则是受体_2_配体。结合能的结果在txt文件里,分子位置的结果在pdbqt文件里。
四、利用python查看和筛选结果
txt文件太多了,需要批量的查看和处理,这里我选用了python。因为之前发过一篇结果利用python处理结果的(点我看)。阅读量竟然是最高的,也是令人震惊。所以这里也还用python,最后绘制个热图,好看一点。
首先,载入所需的包
import pandas as pd
import numpy as np
import os
定义获取结合能的函数
def getmaxaffinity(fpath):
m = []
try:
with open (fpath, encoding = "utf-8") as f:
for i in f.readlines()[-10]:
if i!= ' ' and i != '\n':
m.append(i)
return ''.join(m[1:5])
except:
return ''
设置文件路径,也就是对接结果存放的results文件夹的路径,读取其中的txt文件。
f = r'C:\\Users\\O_O\\Desktop\\dock\\results'
receptors = []
ligands = []
affinity = []
for root,dirs,files in os.walk(f):
for file in files:
if os.path.splitext(file)[1]== '.txt':
try:
fn = os.path.splitext(file)[0]
receptor,ligand = fn.split('_2_')
receptors.append(receptor)
ligands.append(ligand)
fpath = os.path.join(f,file)
affinity.append(getmaxaffinity(fpath))
except:
continue
这里利用了结果命名的规则,获得受体和配体的信息。所以在对受体和配体的进行命名的时候尽量不要出现
_2_这个字符。
(其实出现也没事,把批处理的命名规则改一下就行,代码小白忽略括号内的内容)
将受体、配体和结合能的信息做出表格以供筛选
df = pd.DataFrame()
df['receptor'] = receptors
df['ligand'] = ligands
df['affinity'] = affinity
数据整理一下,主要是将结合能变成数字。
df['ligand'] = df['ligand'].str.split('_',expand = True)[2]
df = df.replace('', np.nan)
df = df.dropna(axis = 0)
df['affinity'] = df['affinity'].astype(float)
筛选<-0.7的结果
df_h = df.loc[df['affinity'] < -7.0]
五、绘制热图
载入绘图所需要的包
import matplotlib
import matplotlib.pyplot as plt
import seaborn as sns
将对接结果变成一个宽数据以便绘图
df_a = df_h.pivot(index = 'ligand', columns = 'receptor', values = 'affinity')
设置绘图参数,绘图并保存
ax = sns.heatmap (df_a,cmap = 'OrRd',annot = True)
ax.tick_params(top=True, bottom=False,
labeltop=True, labelbottom=False)
heatmap = ax.get_figure()
heatmap.savefig("heat.png",dpi = 300)
结果如图所示,数据有点少。。
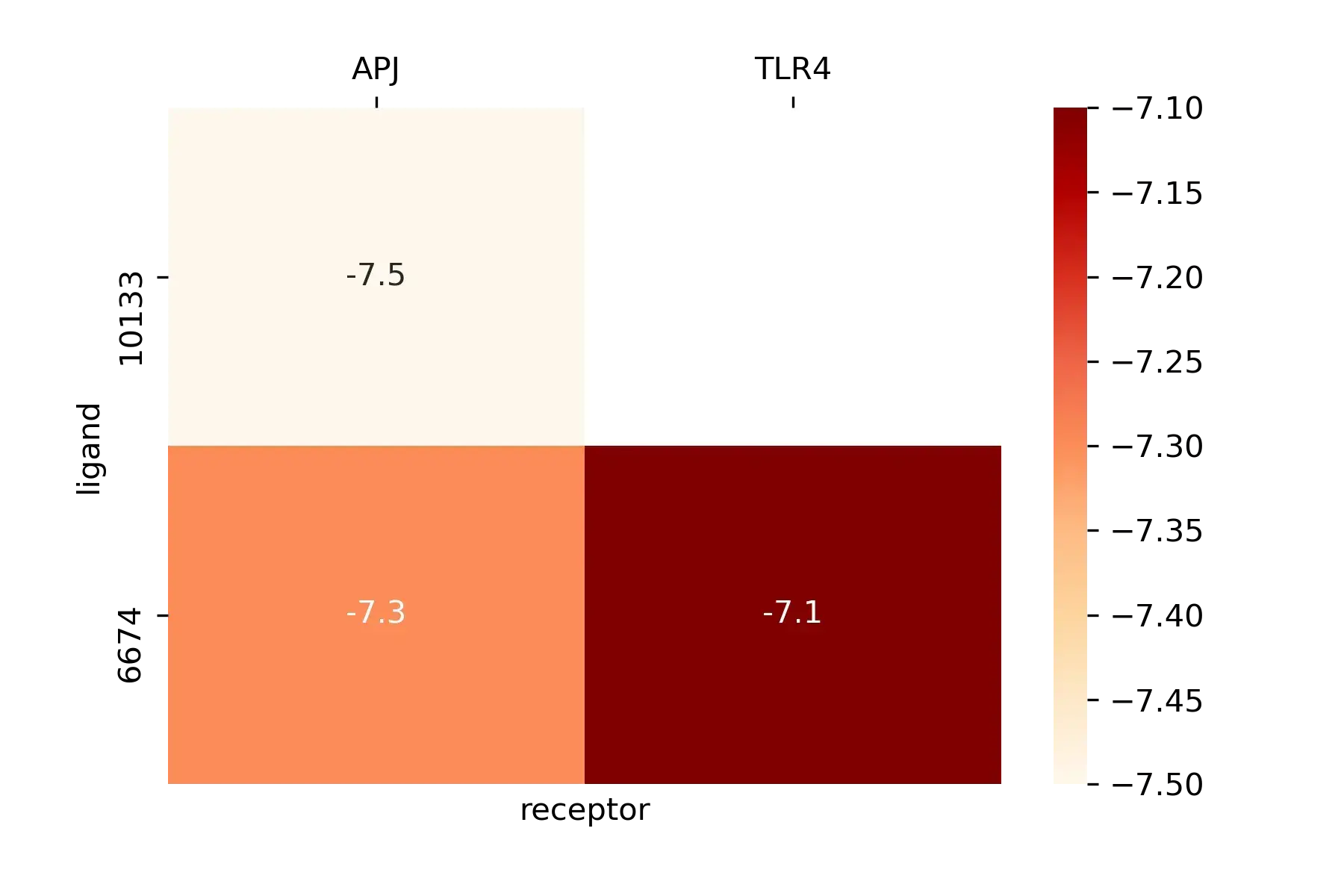
图还可以更好看,只是懒得搞了,需要的话再查吧。
感谢大家看到最后。我也不是专业学编程的,只是最近在搞分子对接就找了一些偷懒的方法把他搞得简单点。写的不好大家多多见谅。最后有什么问题可以大家一起交流,一起学习,共同进步!
不想敲代码的可以直接下载写好的py文件(其实也就是把上面的代码复制下来)。点这里下载
文章出处登录后可见!