目录
软件下载
使用Autodock进行蛋白和小分子复合体的对接需要使用到如下工具,很巧的是,这几个工具都是使用python进行编写的:
pymol:三维结构可视化
https://pymol.org/2/
选择教育版下载即可,若需要license激活,只需注册一个账号即可获得使用许可,不激活也可以正常使用
openbabel:不同三维结构格式文件间转换
https://openbabel.org/wiki/Category:Installation
Autodock:进行蛋白-小分子对接,该文件下载后解压得到的是两个exe文件(Windows版),这两个exe就是最终需要使用的部分,没有什么点击这两个exe然后一路下一步安装的过程
这两个exe放在哪个目录下随意,但路径中一定不能有中文,否则运行过程中会报错!!!
https://autodock.scripps.edu/download-autodock4/
mgltools:Autodock的可视化工具,安装路径中同样不能有中文
https://ccsb.scripps.edu/mgltools/downloads/
以上四个除了Autodock直接解压即可外,其余三个都是直接不断下一步即可,没有什么要注意的地方
三维结构文件获取
蛋白质三维结构:
对于已经有文献报道的蛋白质三维结构,直接从PDB获取即可;未被实验解析的蛋白质结构则可以通过Alphafold来进行预测,使用方法可以参考笔者之前的博文:
【实用教程】使用AlphaFold2进行蛋白质结构在线预测
小分子三维结构:
使用PubChem搜索需要的小分子,在对应结果的structure处下载即可,这里推荐下载sdf格式文件
下载得到小分子三维结构后,使用openbabel将sdf格式文件转换为pdb格式文件
openbabel的使用逻辑非常简单粗暴,左右两边分别选择好输入输出文件的格式和对应路径,然后点中间的CONVERT即可:

Autodock使用
打开Autodock
首先打开Autodock,mgltools下载完毕后是会自动创建几个桌面快捷方式的,我们需要使用到的只有这一个

双击打开后,首先会弹出一个黑色窗口,这个窗口在运行autodock的过程中由始至终都会存在,注意不要关闭,否则将会自动关闭autodock
注意到了这个界面的时候不要傻等着,点一下remind me later,当然你立马注册也可以(autodock的初始界面图是有好几张的,如果这里你的图跟我的不一样不用怀疑自己搞错了)

将蛋白质pdb文件和小分子pdb文件放在同一个目录下,注意路径中不能包含中文!!! 将软件运行目录设置在蛋白质和小分子所在目录中


在All Molecules处点击右键,选择Read molecule,打开蛋白质pdb文件

去水:
随后需要将pdb文件中的水分子移除(该操作只能移除水分子,如果pdb文件中还有其他小分子的建议使用pymol手动删除)
注意:只有从PDB数据库中下载的实验结构才会包含水分子或其他小分子,使用alphafold预测的蛋白质结构不包含水分子,可跳过该步
Select->Select From String


配体小分子准备
打开配体小分子pdb文件

注意这里文件默认后缀名是.pdbqt,我们输入的是.pdb文件,需要选择一下可用文件类型,all files或者.pdb都可以

因为这时候还维持在大分子的视图,所以小分子是很小的,如果想看到小分子的话可以把大分子的L的红圈给点掉(此步非必须)

接下来需要设置小分子可以如何扭曲。首先需要检测扭曲中心:

点击后将小分子放大可以看到有一个原子被绿色球所包裹,该原子即为中心
按住左键移动鼠标为旋转;按住鼠标右键移动鼠标为放大缩小;按住鼠标中键即滚轮移动鼠标为移动位置

通过如下方法可以设置分子中哪些键是可以扭曲的

扭曲规则是根据化学性质来的,当然你可以可以根据个人需要设定哪些键可以扭曲哪些不能扭曲,如果你啥也不懂那就不用动了。选择完毕后就Done即可。

接着设置扭曲原子的数目:

我个人倾向于选择最大扭曲数,当然这里可以扭曲的原子越多后面需要消耗的计算资源也越多,时间越长。设置好后Dismiss就行。

将设置好的小分子保存为.pdbqt文件:

蛋白质大分子的分割
在这一步中我们需要选择目标蛋白质区域进行分子对接。

选择你的大分子,然后Select Molecule,如果他后面有问你一堆乱七八糟的东西你就Yes就好

然后打开Grid Box,选择目标蛋白质区域

如果你先前已经对蛋白质有一定的了解,知道分子对接可能的作用口袋,那你只需要选择该口袋进行对接就可以了。如果不了解的话选择全蛋白即可。
如果先前有把蛋白那个L的红色点点掉的话可以重新点亮,并缩小整个视图,使你能看到整个完整的蛋白。
图中红绿蓝三色的正方体所包围区域即为需要对接的蛋白质区域
通过调整xyz的大小、整体倍数(下图红框)和xyz中心来覆盖目标区域

选择完毕后点击关闭并保存(不要直接右上角×出去了)

再次确定小分子文件(我也不懂这步意义是啥,但就照做吧……)

输出grid设置文件。建议保存位置就在跟大分子和小分子同一目录下,不要放其他地方。路径不能有中文。同时在输出文件名的时候记得手动给他把后缀名.gpf加上,他自己不会自动加后缀名……

运行AutoGrid 4
选择运行AutoGrid 4:

找到你之前解压的autogrid4.exe,并找到上一步保存的==.gpf文件==

如果这两个文件都能顺利找到后第三行会自动填充,此时点Launch即可。如果前面的.gpf文件没有后缀名这里可能会出错。
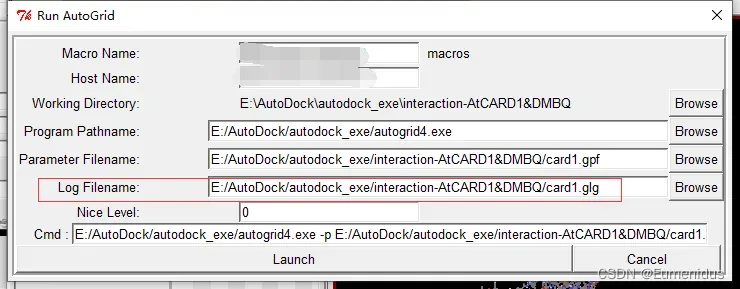
开始运行后会出现这样一个小窗,说明程序正在运行,小窗消失说明运行完成。
如果你的这个小窗在十秒内消失了那你可能要反思一下是不是之前有哪步做错了,比如在刚开始的时候软件的运行路径设置错了,没有设置在大小分子的目录下。
进行分子对接
需要再次设置刚性分子文件,选择大分子蛋白的.pdbqt文件打开:

选择小分子配体文件,弹出的窗口accept就行:

选择对接算法,我个人一般会用遗传算法,读者也可以自行尝试其他算法:

在弹出的窗口中修改Number of GA Runs,这里指的是后面要重复对接多少次,次数越多消耗时间越长,结果相对更可靠,这个数字一般不要低于100,设置好后accept:

设置完毕后输出.dpf文件,注意这里选择的是Lamarckian GA而不是刚刚的Genetic Algorithm。
跟gpf一样,dpf的后缀名也需要自己加一下。

运行AutoDock4
选择运行AutoDock4:

同样地,设置好autodock4.exe的路径和.dpf文件路径,log filename一行即可自动生成,然后Launch

同样会出现一个小窗,小窗消失即运行完成,前面设置的重复次数越多这一步越耗时

对接结果查看及评估
打开对接结果.dlg文件

选择大分子文件:

查看对接后小分子结果,通过点击弹出的小窗(下图上方)的左右箭头即可看到小分子位置,如果你觉得点了几下没看到变化那是小分子太小你没看到……

对对接结果构象进行聚类分析:

该柱状图表示某一类构象有多少个相似的对接结果,如果在数次重复中小分子都对接到了同一口袋即使小分子扭曲构象略有区别,也会被聚类到一起,纵轴表示频数,数目越多表示该构象越稳定,横轴则是能量,能量越低越稳定
像这里展示的这个构象如果在对接次数足够多的情况下依然每种都只有一个那就说明对接结果不太可靠
(因为Autodock功能比较简单,对接结果较差不能说明该分子与蛋白就一定没有互作)
单击柱状图某一根柱子该柱子会变为红色,相应构象也会显示

可以选择合适的构象进行保存,输出的文件记得加后缀名.pdbqt,使用Openbabel转为.pdb文件,可以使用pymol对.pdb文件进行可视化,并进一步分析相互作用:

使用PLIP分析相互作用
使用PLIP分析对接结果,该网站使用方法非常简单,只需要直接输入对接结果的pdb文件,然后点击Analyze:

在输出结果中,会展示包括氢键等各种常见的相互作用力,并注明蛋白质中具体哪个氨基酸残基与小分子互作,方便用户后续进行点突变等操作:

结语
AutoDock总体来说还是比较像是一款玩具,只能解决很简单的分子对接问题,同时也只能在个人计算机电脑上进行单个操作,如果需要专业的对接还需要用服务器进行大规模的筛选。
最后关于本教程,由于文章较长且本人一直摸鱼,实际写的过程跨度了几个月,中间有些步骤比如发现怎么我的小分子/蛋白又消失了出现了跟你的不太一样,不用担心,那是因为我中间关了又重开软件。
文章出处登录后可见!