单倍型块( haplotype block )是连续基因组区域的一系列遗传变异位点的组合,这种基因组区域通常表现为高连锁不平衡、低重组。
但其实在育种过程中,情况可能更为复杂,我们观察到的haplotype block到底是由育种家的人工选择维持的(比如控制多性状的基因组区域,育种家会聚合多种优良的性状,从而形成单一的单倍型),还是由于该染色体区域重组率低而不易被打破,不同的研究似乎有不同的见解。有研究表明,一些聚合了多性状且形成了haplotype block的基因组区域,通过研究后代的重组自交系,表明这些haplotype blocks能够被打破 。当然,这并不能否认一些高连锁不平衡、低重组的基因组局域对维持haplotype blocks的作用,并且也确实观察到这样的现象。或许无论是人工选择还是染色体本身的生物学特性,两者并不冲突,可能在维持haplotype block均发挥了作用。
HaploBlocker是一个R包,在群体遗传分析中,用于寻找haplotype block (单倍型块)。根据官方文档,HaploBlocker包括八个部分的参数,分别为:1)Prefilters(SNP数据集的预处理),2)Cluster-building,3)Cluster-merging,4)Block-identification,5)Block-filtering,6)Block-extending,7)Off-variant-identification(optional),8)Performance parameters – computing time。
这里我们主要详细介绍前面六个部分的参数。更多详细信息请参考:https://github.com/tpook92/HaploBlocker/wiki/
1)Prefiters
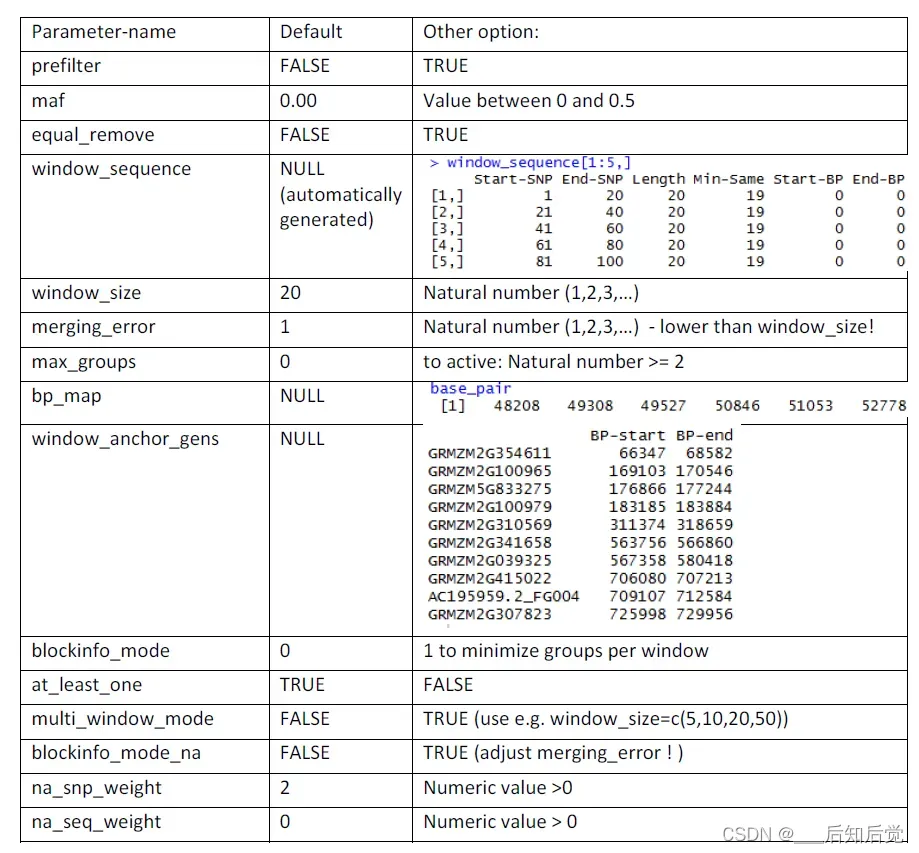
Parameters: prefilter, maf, equal_remove
maf:最小等位基因频率
equal_remove:one can remove all SNPs in perfect LD to the previous one (该参数的官方文档解释,未能理解其中含义)
prefilter:默认情况下不进行SNP过滤,可通过prefilter参数进行激活
2)Cluster-bulding
Parameters: window_sequence, window_size, merging_error, max_groups, bp_map, window_anchor_gens, blockinfo_mode, at_least_one, multi_window_mode, blockinfo_mode_na, na_snp_weight, na_seq_weight, actual_snp_weight
window_size:窗口大小
merging_error:在每个窗口中允许出现相同数量的错误
window_sequence:如果想使用不同的窗口大小和每个区域的错误数量 (例如,准确地跨越窗口根据基因的位置),可以通过参数手动设置
bp_map:要包含碱基对中的位置,必须通过该参数输入每个SNP的位置。
window_sequence:生成window_sequence
max_group:用于选择窗口边界,以获得每个窗口一定数量的变异(当前一个block的变异比max_groups多时,就会启动下一个窗口)
window_anchor_gens:当提供所需窗口边界的物理位置(例如基因的起始/结束点),在参数window_anchor_gens中提供它们,得到的window_sequence会自动计算出来。注意,不支持重叠窗口。
blockinfo_mode:为了减少每个窗口中的group的数量(默认情况下,group是根据window中最常见的单倍型衍生的)
at_least_one:一个实用程序参数,用于确保每个窗口中至少有一个SNP必须是相同的(仅与window_size merging_error相关)
multi_window_mode:设置为TURE,在拟合过程中使用多个window clusters。window_size,merging_error,和min_share能够将vector作为输入处理,和/或window_sequence可以作为不同窗口序列的列表。每个元素都被单独处理。在没有提供vector/list的情况下,输入用于所有情况。
block_mode_na,na_snp_weight,actual_snp_weight:在数据集包含缺失值的情况下,默认值将被建模为分析中的另一个等位变异 (“9”)。如果统计NAs和具有不同权重的等位变异之间的差异,可以激活block_mode_na。NA和等位变异之间的差异被计数为na_snp_weight合并错误,而不同的等位变异被计数为actual_snp_weight合并错误。在标记只包含一个等位变异的情况下,NAs差异被计算为na_sep_weight合并错误。需要注意的是,这种模式非常耗费时间,而且仍然可以进行一些更改。请注意,工具所需的输入是单倍型,所以在应用之前需要进行phasing。
3)Cluster-merging
Parameters: node_min, gap, min_reduction_cross, min_reduction_neglet, early_remove, node_min_early
node_min:在cluster-merging时,每个node的单倍型数量可以通过node_min来控制。
gap:To avoid short segments between removed nodes, all haplotypes are in a common variant for less than gap windows are removed from the window cluster.
min_reduction_cross,min_reduction_neglet,early_remove,node_min_early:为了减少SG,SM和NN,SG,SM, SG循环的计算时间,可以使用min_reduction_cross和min_reduction_neglet来停止循环,当在一个循环的算法中发生的合并数小于这个数。如果节点数量多,单倍型少,可以考虑在SG、SM周期之前通过early_remove和node_min_early删除它们。
4)Block-identification
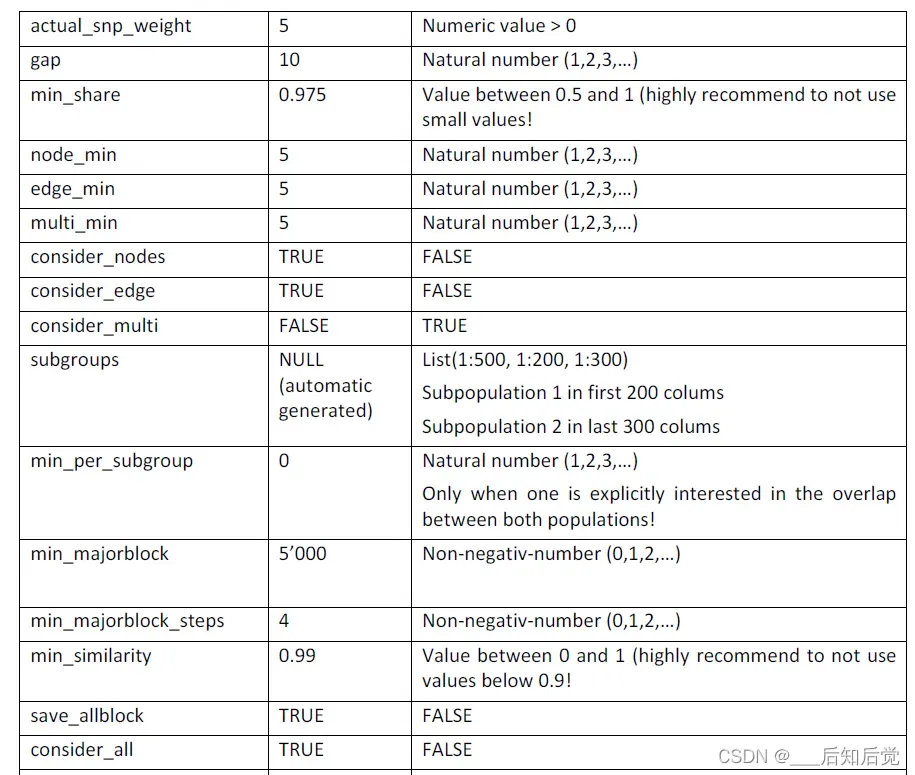
Parameters: min_share, subgroups, consider_nodes, consider_edge, min_per_subgroup, consider_multi, multi_min, node_min, edge_min, double_share
consideration _nodes,consideration _edge:如果在识别步骤中不考虑节点(nodes)或边界(edges)作为起始blocks,可以将consideration _nodes和consideration _edge设置为FALSE。这两种更改都不推荐(将其中一个设置为FALSE将减少计算时间)。
consider_multi:基于两个相邻边的单倍型筛选块,使用consider_multi(仅推荐用于小数据集,并首先使用multi_window_mode)
multi_min, node_min, edge_min:要更改每个块的最小单倍类型数量,可以使用edge_min (Blocks by Edge)、node_min (Blocks by node) 和 multi_min (multiple edges)
min_share:如果改变扩展block所需要的同一节点中块转换的最小比例,可以使用min_share参数。可以控制每个区块的平均长度和来自一个区块的单倍型之间的相似性。数值越高,区块越短,因此,一个block的单倍型之间的相似性更高,总体的block数量也更多。此外,overlapping blocks的数量也大大减少。
subgroup,min_per_subgroup:如果需要形成不仅适用于整个数据集,而且适用于subgroups的blocks,可以使用参数subgroup,并设置每个block中每个subgroup的最小数量的单倍型(min_per_subgroup)。该参数的更改会导致数据集中所有subgroup中的blocks,因此可能导致低覆盖率(coverages)。只有当明确地对多个subgroups的重叠区域感兴趣时,才建议在这里进行更改。
double_share:在block-identification(extended-blockidentification)中同时考虑长片段和短片段,将double_share设置在同一较长的片段中转变所需的单倍型的最小比例。
5)Block-filtering
Parameters: min_majorblock, min_majorblock_steps, min_similarity, save_allblock, consider_all, merge_closeblock, max_diff_i, max_diff_l, off_lines, weighting_length, weighting_size, target_coverage, target_stop
min_majorblock:主要的筛选过程是通过识别单元格的数量来完成的,其中每个单元格是数据集中最相关的单元格。这个数字可以通过min_majorblock来改变,并且应该用来在块的数量和块库的覆盖率之间找到一个平衡。
target_coverage:如果需要获得具有特定覆盖率的单倍型库,建议使用target_coverage参数来初始化一个自动拟合过程,以确定min_majorblock的一个好的选择。
max_iteration,min_step_size,target_stop:如果需要控制在target_coverage中适合min_majorblock的迭代次数,使用max_iteration和min_step_size控制每步min_majorblock的最小差异,target_stop为目标提供最大差异。
weighting_length,weighting_size:如果需要控制每个单元格中哪个block是最相关的block,可以使用参数weighting_length和weighting_size来控制每个block中单倍型的长度和数量之间的权重。
min_majorblock_steps:避免排除重要的块,最小数量缓慢增加(在min_majorblock_steps线性增加的步骤中)。
min_similarity,save_allblock:单倍型与包含块的最小相似度可以通过参数min_similarity来设置。该参数可以控制同一block的两个单倍型之间的最小相似性。单倍型不满足min_similarity,但在所有用于识别块的节点中不会被删除,除非参数save_allblock设置为FALSE。
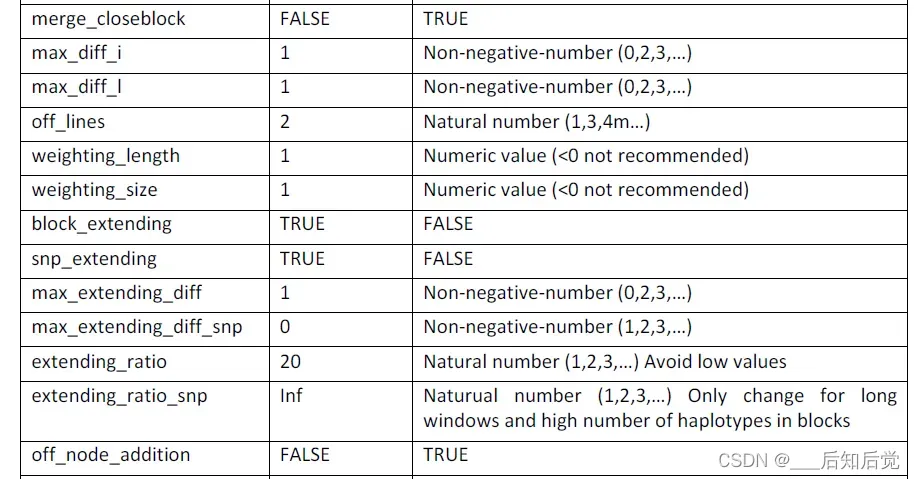
过滤过程中还有一些小参数:
consideration_all:如果不考虑不在block中的单倍型,必须将consideration_all设置为FALSE。
merge_closeblock,max_diff_i,max_diff_l:如果允许具有类似单倍型和位置的block被合并,必须激活merge_closeblock并通过max_diff_i(不同单倍型)和max_diff_l(两者之间的差异)设置它们之间的最大差异。
off_lines:当window sequence相同时,一个block必须与另一个block相比较的最小附加单倍型数可以通过off_lines来控制。
6)Block-extending
block_extend,snp_extend:如果不希望执行block和SNP扩展,则将block_extend和/或snp_extend设置为FALSE。
max_extending_diff,max_extending_diff_snp,max_extending_diff,max_extending_diff_snp:如果想要使用扩展,可以控制在某些单倍型(max_extending_diff,max_extending_diff_snp)中不同的window的最大数量,以及有和没有变化的window之间的比例(extending_ratio,extending_ratio_snp)。
典型输入
HaploBlocker典型参数设置
接下来,我将介绍如何具体利用HaploBloker软件进行单倍型block分析。
文章出处登录后可见!