人工智能到深度学习:药物发现的机器智能方法
Artificial intelligence to deep learning: machine intelligence approach for drug discovery
Rohan Gupta1、Devesh Srivastava1、Mehar Sahu1、Swati Tiwari1、Rashmi K.Ambasta1、Pravir Kumar1
接收日期:2021年1月29日,接受日期:2021年3月22日,在线发布日期:2021年4月12日
©作者,根据Springer Nature Switzerland AG 2021的独家许可
Rohan Gupta、Devesh Srivastava、Mehar Sahu和Swati Tiwari对这项工作做出了同等贡献。
通讯作者:Pravir Kumar
pravirkumar@dtu.ac.in;kpravir@gmail.com
1印度,德里110042,Bawana路,Shahbad Daulatpur,德里科技大学生物技术系(原DCE),分子神经科学和功能基因组学实验室
摘要
药物设计与开发是制药企业和化学科学家研究的重要领域。然而,低疗效、靶向给药、时间消耗和高成本给药物设计和发现带来了障碍和挑战。此外,来自基因组学、蛋白质组学、微阵列数据和临床试验的复杂和大数据也给药物发现管道带来了障碍。人工智能和机器学习技术在药物发现和开发中起着至关重要的作用。换句话说,人工神经网络和深度学习算法已经使这个领域现代化。机器学习和深度学习算法已应用于多肽合成、基于结构的虚拟筛选、基于配体的虚拟筛选、毒性预测、药物监测和释放、药效团建模、定量构效关系、药物重定位、多重药理学和理化活性等药物发现过程。过去的证据加强了人工智能的实施和这一领域的深入学习。此外,新的数据挖掘、管理和管理技术为最近开发的建模算法提供了关键支持。综上所述,人工智能和深度学习的发展为合理的药物设计和发现过程提供了极好的机会,最终将影响人类。
图形摘要
与药物设计和开发相关的主要问题是时间消耗和生产成本。此外,效率低下、靶向给药不准确和剂量不当也是阻碍药物递送和开发过程的其他障碍。随着技术的进步,集成人工智能算法的计算机辅助药物设计可以消除传统药物设计和开发的挑战和障碍。人工智能被称为包含机器学习的超集,而机器学习包括有监督学习、无监督学习和强化学习。此外,深度学习作为机器学习的一个子集,在药物设计和开发中得到了广泛的应用。人工神经网络、深度神经网络、支持向量机、分类与回归、生成对抗网络、符号学习和元学习是应用于药物设计和发现过程的算法示例。人工智能已应用于药物设计和开发过程的各个领域,如从多肽合成到分子设计、虚拟筛选到分子对接、定量构效关系到药物重新定位、蛋白质错折叠到蛋白质相互作用、分子通路识别到多重药理学等。人工智能原理已被应用于活性和非活性的分类、药物释放监测、临床前和临床开发、一级和二级药物筛选、生物标志物开发、药物制造、生物活性鉴定和理化性质、毒性预测和作用方式识别。
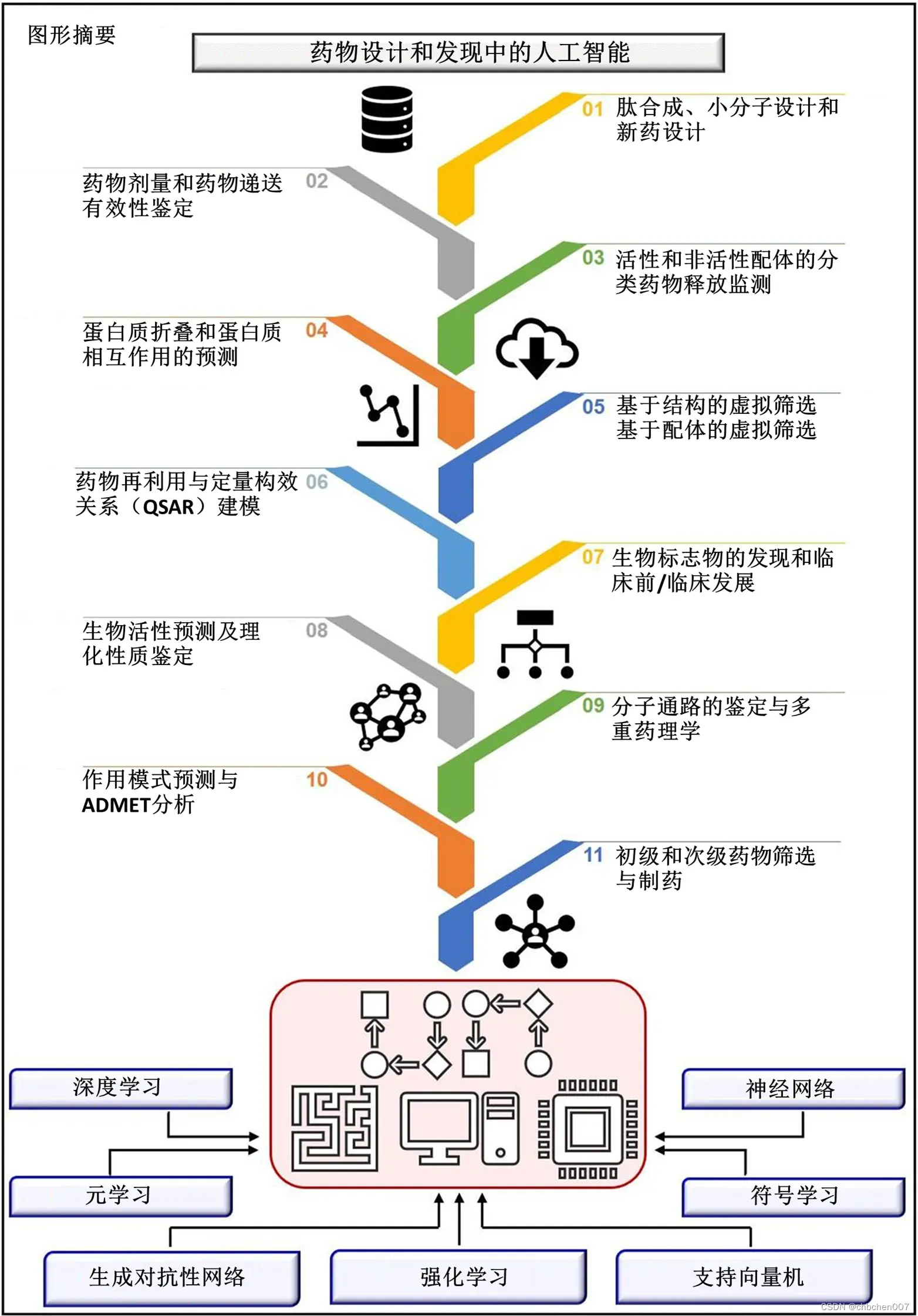
关键词:人工智能、机器学习、深度学习、虚拟筛选、药物设计与发现、人工神经网络、计算机辅助药物设计、定量结构-活性关系、药物再利用
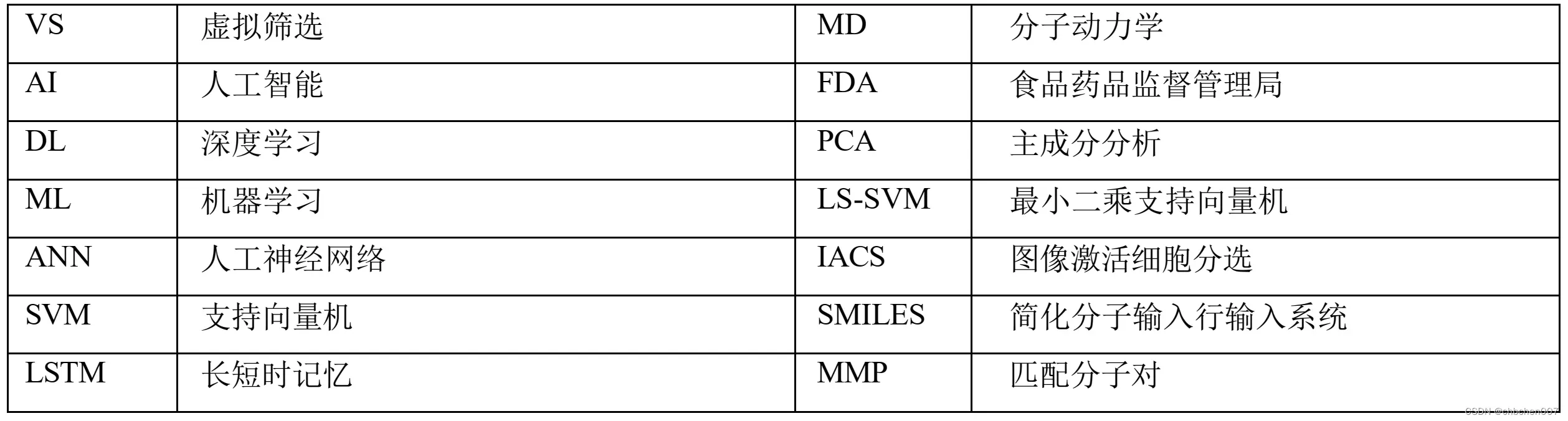
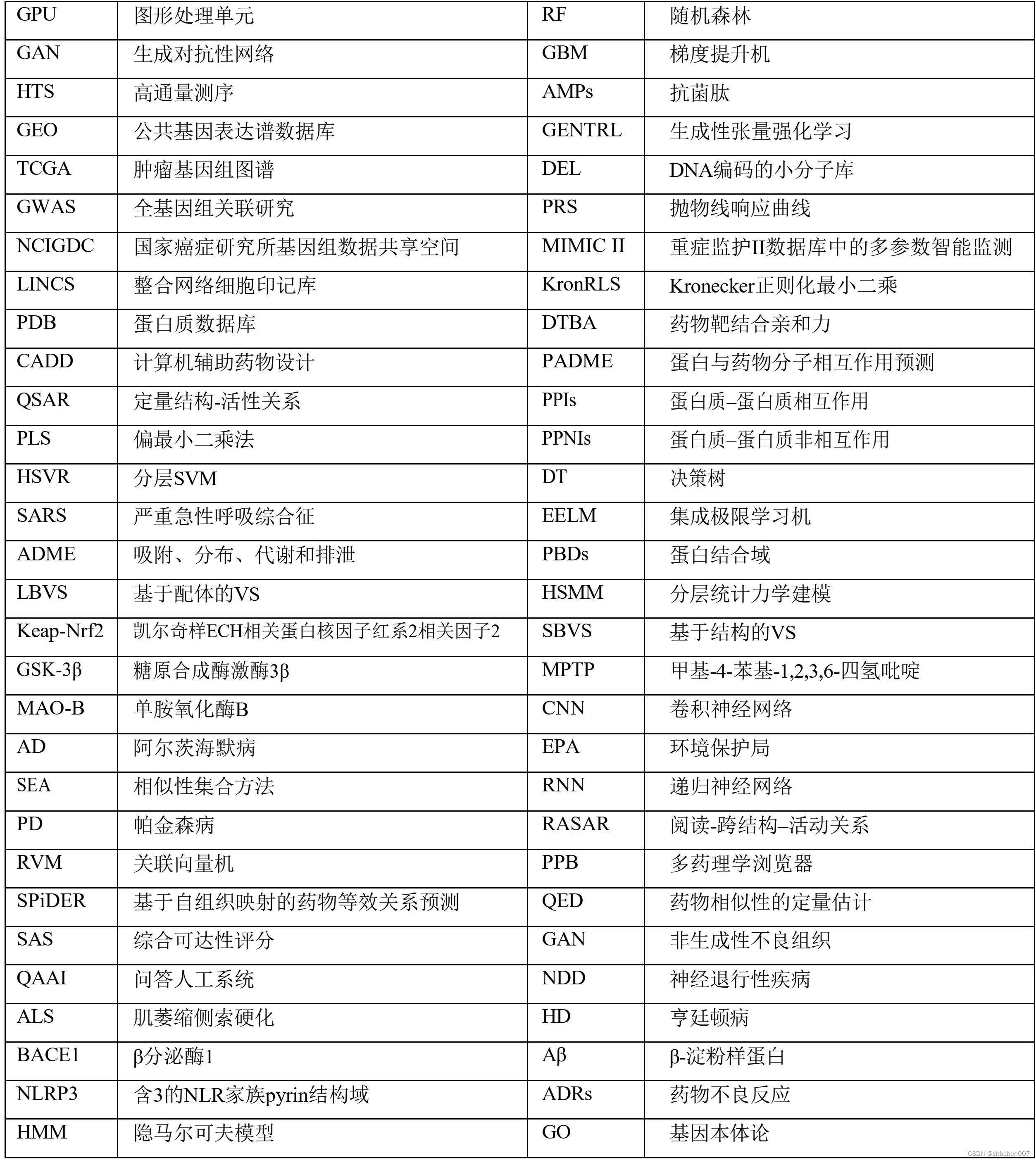
缩写词
1. 简介
在过去的二十年中,高效、先进的治疗药物靶向给药系统的发展给化学和生物科学家带来了巨大的挑战[1]。此外,开发新型治疗药物的开发成本和时间消耗是药物设计和开发过程中的另一个挫折[2]。为了最大限度地减少这些挑战和障碍,世界各地的研究人员转向计算方法,如虚拟筛选(VS)和分子对接,也被称为传统方法。然而,这些技术也带来了诸如不准确和效率低下等挑战[3]。因此,新技术的实现出现了激增,这些新技术能够自给自足地消除传统计算方法中遇到的挑战。人工智能(AI),包括深度学习(DL)和机器学习(ML)算法,已经成为一种可能的解决方案,它可以克服药物设计和发现过程中的问题和障碍[4]。此外,药物发现和设计包括漫长而复杂的步骤,如靶点选择和验证、治疗性筛选和先导化合物优化、临床前和临床试验以及生产实践。所有这些步骤都给确定有效的药物治疗疾病带来了另一个巨大的挑战。因此,摆在制药公司面前的最大问题是管理成本和过程速度[5]。人工智能以一种简单、科学的方式回答了所有这些问题,减少了过程的时间消耗和成本。此外,制药公司和医疗保健部门数据数字化的增加推动了人工智能的实施,以克服审查复杂数据的问题[6]。
人工智能也称为机器智能,是指计算机系统从输入或过去的数据中学习的能力。当机器在学习和解决问题的过程中模仿与人脑相关的认知行为时,通常使用人工智能这个术语[7]。目前,生物和化学科学家广泛地将人工智能算法应用于药物设计和发现过程中[8]。基于AI和ML原理的计算建模为化合物的识别和验证、靶点识别、多肽合成、药物毒性和理化性质评估、药物监测、药物疗效和有效性以及药物重新定位提供了重要途径[9]。随着人工智能原理以及ML和DL算法的出现,对化学库中的化合物进行VS变得简单且时效,化学库中包含超过万亿(10^6 million)种化合物。此外,AI模型消除了因靶外相互作用而产生的毒性问题[10]。在此,我们简要讨论AI从ML到DL的演变以及大数据参与药物发现过程的革命性变革。随后,综述了人工智能与常规化学在药物发现过程改进中的融合以及人工智能在传统药物发现过程改进中的应用。随后,我们讨论了人工智能在药物设计和发现过程中的许多应用,如一级和二级筛选、药物毒性、药物释放和监测、药物剂量有效性和疗效、药物重新定位、多药理学以及药物-靶点相互作用
2. 人工智能的进化:机器学习到深度学习
2015年9月,Google搜索趋势显示,ML引入后,AI是搜索最多的词。一些人将ML描述为主要AI应用程序,而另一些人将其描述为AI的子集[11,12]。AI是一个总括性术语,其中计算机程序能够像人类一样思考和行为,而ML则超出了将数据输入机器的范围,以及像朴素贝叶斯(Naive Bayes)、决策树(decision tree,DT)、隐马尔可夫模型(hidden Markov models,HMM)等算法,这些算法有助于机器学习而无需显式编程。后来,随着神经网络的发展,机器可以像人脑一样对输入的数据进行分类和组织,这进一步显示了人工智能的进步。20世纪前后,伊戈尔·艾森伯格(Igor Aizenberg)及其同事在谈到人工神经网络(ANN)时,首次提出了“深度学习”一词。DL是ML的一个子集,而ML本身就是AI的一个子集,因此,演化过程是AI>ML>DL[13,14]。ML要么使用监督学习,其中模型被训练为使用标记数据,这意味着输入已被标记为相应的首选输出标签;要么使用无监督学习,其中模型被训练为使用未标记数据,但从输入数据中查找循环模式[15]。另一些是半监督学习,使用监督和非监督学习的结合;自监督学习是一种特殊情况,它采用两步过程,其中无监督学习为未标记数据生成标签,其最终目的是建立有监督学习模型;强化学习是一种ML,它借助于一个恒定的反馈回路,随着时间的推移对算法进行改进,最后是DL,其中有多层ML算法,被称为大脑启发的算法家族,模仿人脑,但需要较高的计算能力来训练和大数据才能成功[16,17]。ML的起源可以追溯到1943年,当时McCulloch和Pitts发表了一篇题为“神经活动中内在思想的逻辑演算”的文章,他们给出了第一个神经网络的数学模型[19]。Alan M. Turing在1950年发表的开创性论文[18]中提出了ML的概念。1952年,Arthur L. Samuel为IBM编写了一个棋盘格程序[20],从而推广了“机器学习”这个术语。1957年,Frank Rosenblatt开发了用于图像识别的感知机[21]。Henry J. Kelley在1960年开发了连续反向传播模型,Stuart Dreyfus在1962年开发了一个仅基于链式规则的简单版本[22,23]。1965年,Ivakhnenko和Lapa开发了第一个可用的DL网络。1980年左右,福岛国彦(Kunihiko Fukushima)开发了一种名为神经认知机的人工神经网络,这种网络具有多层设计,可以帮助计算机学习如何识别视觉模式[24]。他还开发了第一个基于动物视觉皮层组织的卷积神经网络(CNN)[25][图1]。
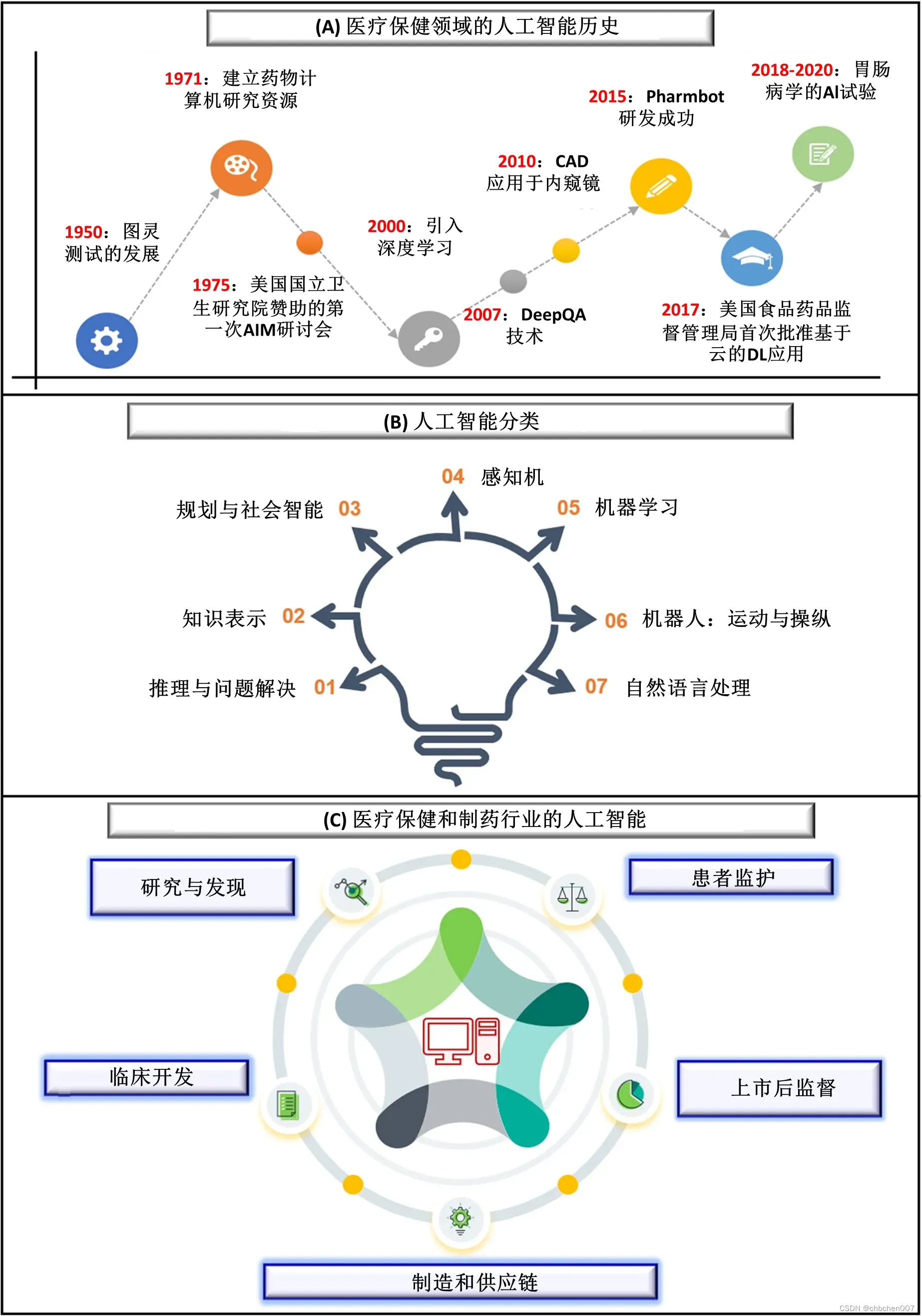
图1 (A)人工智能在医疗保健中的历史:1950年,随着图灵测试的发展,人工智能在医疗保健中的首次突破。后来,在1975年,第一个关于计算机在医学上的研究资源被开发出来,随后NIH的第一个以AIM为中心的研讨会标志着人工智能在医疗保健中的重要性。随着21世纪深度学习的发展和2007年DeepQA的引入,人工智能在医疗领域的应用范围不断扩大。此外,2010年,CAD首次应用于内镜检查,而2015年,第一台Pharmbot被开发出来。2017年,FDA批准的第一个基于云的DL应用程序被引入,这也标志着人工智能在医疗领域的实施。从2018年到2020年,在胃肠病学领域进行了多个AI试验。(B)人工智能分类:有七种人工智能分类,即推理和问题解决、知识表示、规划和社会智能、感知、机器学习、机器人运动和操纵以及自然语言处理,正如Russel和Norvig在其《人工智能:现代方法》一书中所讨论的那样。机器学习进一步分为三个重要的子集:有监督学习、无监督学习和深度学习,而视觉又分为两个子集,如图像识别和机器视觉。类似地,语音被划分为两个子集:语音到文本和文本到语音,而自然语言处理被划分为五个主要子集,包括分类、机器翻译、问答、文本生成和内容提取。人工智能在医疗和制药行业有五个重要的应用,它们改变了整个场景。这些应用包括研究和发现、临床开发、制造和供应链、患者监测和上市后监测
David Rumelhart、Geoffrey Hinton和Ronald J. Williams于1986年发表了一篇题为“通过反向传播错误学习表征”的论文,该论文证明反向传播可以改善形状识别和单词预测[26]。在最初的成功之后,也有一些挫折,但辛顿在第二个人工智能冬季继续努力,以达到新的高度。因此,他被认为是DL的教父。很快,在1989年,Yann LeCun在贝尔实验室首次进行了反向传播的实际演示[27]。同年,Christopher Watkins发表了题为“从延迟奖励中学习”的论文,该论文引入了Q-Learning的概念,进一步改进了计算机程序中的强化学习[28]。1995年,Corinna Cortes和Vladimir Vapnik开发了支持向量机(SVM)来映射和识别相似的数据[29]。两年后的1997年,Jürgen Schmidhuber和Sepp Hochreiter为递归神经网络开发了长-短期记忆(LSTM)[30]。
1999年,图形处理单元(GPU)作为微处理器电路推出,最初是为了加速计算机游戏的三维图形处理而开发的。后来,GPU由于具有并行计算能力而成为技术和研究领域的热点。META Group在2001年发布的一份研究报告中指出,数据的数量、速度、来源和类型都在不断增加,这是对大数据进军的一种呼唤。2007年,Nvidia引入了计算统一设备架构(CUDA),这是一个允许程序员和研究人员使用GPU进行通用计算的框架[31]。从那时起,在CUDA的帮助下,研究人员开始使用GPU进行DL驱动的操作,因为GPU的高内存带宽允许轻松处理DL算法中涉及的大量数据,GPU中的数千个内核允许同时并行处理神经网络。2009年,李飞飞推出了ImageNet,这是一个免费数据库,包含数百万张可用于研究目的的标记图像 [32]。AlexNet是一种卷积神经网络,由Alex Krizhevsky于2012年左右创建,它使用校正的线性单元来帮助增强速度和落差[33]。同年,Google Brain进行的“猫实验”得出结论,该网络能够正确识别不到16%的呈现对象[34]。2014年,Nvidia引入了CUDA深度神经网络(cuDNN),这是一个基于CUDA的DL库,加速了基于DL的运行[35]。同样,2014年开发并发布了“Deep Face”,用于识别人脸,准确率为97.5%[36]。同年引入了产生式对抗网络(generative adversative networks,GANs),使用两个相互竞争的神经网络来检验数据是否真实或生成[37]。2016年,Cray Inc.在其XC50超级计算机上使用了微软的神经网络软件,该计算机具有1000个Nvidia Tesla P100 GPU,可以执行任务并在几秒钟内输出。2017年,英伟达推出了Tesla V100 GPU,其tensor内核加速了基于AI的运行。然而,DL仍处于成长阶段,在这一领域的进一步发展需要创造性的想法。
3. 变革药物发现过程:大数据和人工智能的作用
大数据可以定义为过于庞大和复杂的数据集,无法用传统的数据分析软件、工具和技术进行分析。大数据的三个主要特征是体积、速度和多样性,其中体积表示生成的大量数据,速度表示这些数据的复制速度,多样性表示数据集中存在的异质性[38]。随着微阵列技术、RNA测序(RNA-seq)技术和高通量测序技术的出现,每天都会产生大量的生物医学数据,当代药物发现也因此向大数据时代过渡。在药物发现中,第一步也是最重要的一步是确定与疾病病理生理学相关的适当靶点(例如,基因、蛋白质),然后找到能够干扰这些靶点的适当药物或类药物分子,现在我们可以访问一组生物医学数据存储库,在这方面可以帮助我们[39]。此外,人工智能的发展使得大数据分析变得更加容易,因为现在有无数的ML技术可用,可以帮助提取这些大型生物医学数据集中存在的有用特征、模式和结构[40]。对于靶点识别,像基因表达这样的特征被广泛应用于理解疾病机制和寻找与疾病相关的基因。微阵列技术和RNA-seq技术为各种疾病提供了大量的基因表达数据。NCBI基因表达综合数据库(GEO)(https://www.ncbi.nlm.nih.gov/geo/)[41]、癌症基因组图谱(TCGA)(https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga) [42]、Arrayexpress(https:// www.ebi.ac.uk/arrayexpress/)[43],是包含基因表达数据的大型存储库。通过分析基因表达特征,我们可以找到导致不同疾病的靶基因。例如,van IJzendoorn等人2019利用ML方法和基因表达数据发现了罕见软组织肉瘤的新生物标记物和潜在药物靶点[44]。
此外,全基因组关联研究(GWAS)可以确定基因组变异与特定复杂疾病之间的相互关系[45]。GWAS central(https://www. gwascentral.org/)[46]、NHGRI-EBI GWAS Catalog(https:// www.ebi.ac.uk/gwas/home)[47]是包含GWAS数据的一些存储库。此外,借助于GWAS,我们可以确定与疾病相关的基因位点,并且已经观察到与这些位点相关的基因是潜在的治疗靶点。例如,Li等人[48]使用GWAS catalog、基因表达、表观基因组学和甲基化数据,通过ML分析确定与幼年特发性关节炎位点相关的靶点基因。此外,其突变可导致不同威胁性疾病的特定基因也是很有希望的治疗靶点。这些风险基因可以通过分析各种基因组和外显子组测序数据来识别。对于测序数据,我们有公共存储库,如Sequence read archive(https://www.ncbi.nlm.nih.gov/sra)[49],其中包含从下一代测序技术获得的测序数据。国家癌症研究所基因组数据共享空间(NCIGDC)(Home | NCI Genomic Data Commons) [50]和TCGA是包含与癌症相关的测序数据的数据库。此外,利用大数据和人工智能,Han等人2019开发了DriverML(https://github.com/ HelloYiHan/DriverML),这是一种基于监督ML的工具,可以指出与癌症相关的驱动基因[51][图2]。
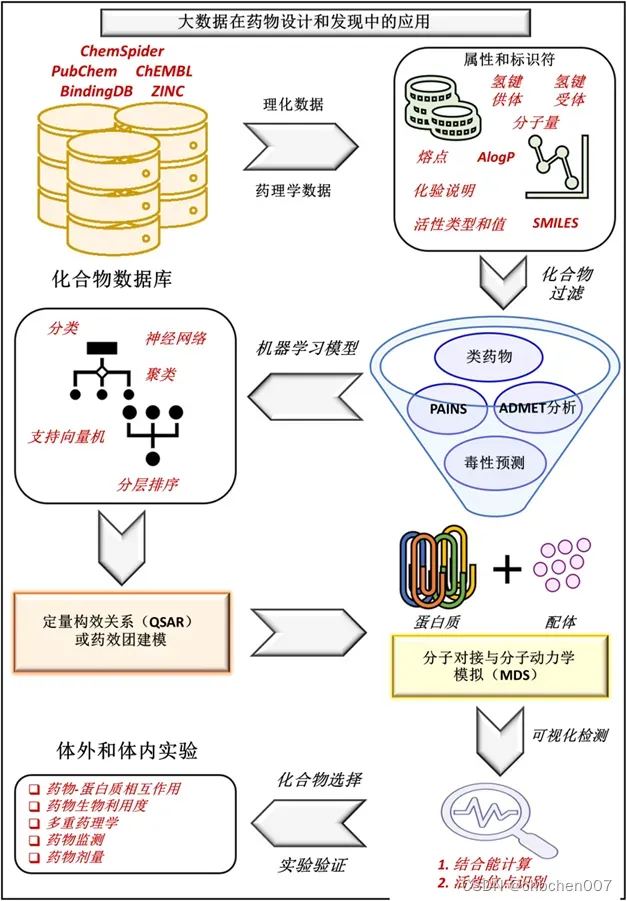
图2 大数据在药物设计和发现中的应用:随着文献、体外、体内、临床研究、基因组学研究、蛋白质组学研究、代谢组学研究、基因本体研究、分子通路数据等生物和化学数据的增加,不同的数据库也在发展。例如,ChemSpider、ChEMBL、ZINC、BindingDB和PubChem是药物设计和发现过程中化合物合成和筛选的基本数据库。对上述数据库中存储的数据进行整理并筛选出药物发现过程所需化合物的药理学和物理化学性质,而不是溶剂化能和质子亲和力、波函数、原子力和过渡态等量子力学计算。高通量筛选数据根据药物相似性、PAINS计算、ADMET分析和毒性进行过滤。对筛选出的化合物进行深度学习、随机森林、分类回归、神经网络等人工智能模型进一步分析。然后对这些化合物进行定量构效关系和药效团模型,然后进行分子对接和分子动力学模拟研究。然后,将最终预测的化合物可视化,用于结合能计算和活性位点识别。因此,最终化合物被鉴定并进行体外和体内实验研究以进行验证。然而,量子力学性质在药物的发现和设计过程中起着至关重要的作用,但这些性质并不能直接阻碍药物的设计过程。QM方法包括从头算密度泛函理论和半经验计算,其中精确计算使用电子关联方法。QM将成为计算药物化学家的重要工具。因此,现代QM方法将在熟悉和简化药物发现过程中发挥更直接的作用
此外,有时甚至已发表的文献也可用于靶点识别,PubMed(https://pubmed.ncbi.nlm.nih.gov/)[52]是各种已发表生物医学文献的主要存储库,其数据挖掘可帮助识别不同疾病的靶点。在确定和验证了合适的靶点之后,下一步是找到合适的药物和/或类药物分子,它们可以与靶点相互作用并引发期望的反应[53]。在大数据时代,我们可以使用大量的大型化学数据库,这有助于找到针对特定靶点的完美药物。同样,PubChem(https:// pubchem.ncbi.nlm.nih.gov/)[54]是一个可自由访问的化学数据库,其中包含各种化学结构的数据,包括其生物、物理、化学和毒性特性[55]。此外,ChEMBL数据库(ChEMBL Database)[56]是一个开放存取的大数据库,包含许多具有药物样特性的生物活性化合物的数据[57]。ChEMBL数据库还包含有关吸收、分布、代谢和排泄(ADME)、这些化合物的毒性特性,甚至其靶点相互作用的信息。此外,DrugBank(https://go.drugbank.com/)[58]是另一个开放存取的药物数据存储库,其中包含各种药物及其靶点和机制的数据[59]。此外,集成的基于网络的细胞特征库(LINCS)L1000(NIH LINCS Program)[60] 是另一个存储库,其中包含了人类细胞系在用不同化合物治疗时基因表达特征变化的信息。LINCS L1000数据驱动搜索引擎,称为L1000CDS^2,是一个开放存取的搜索引擎,其中包含可以恢复差异表达基因表达的药物数据;因此,它们也可用于药物发现[61]。此外,蛋白质数据库(PDB)(RCSB PDB: Homepage)[62]是另一个可自由访问的在线存储库,其中包含蛋白质、DNA、RNA的三维结构数据[63]。PDB数据也被广泛用于评估蛋白质与配体的相互作用,然后找到合适的靶蛋白抑制剂。Xu等[64]结合ML和分子对接寻找新冠病毒3CL蛋白酶抑制剂;本文从PDB中获得了COVID 3CL蛋白酶的晶体结构。
4. 人工智能与常规化学的融合:促进药物发现
在制药工业中,人工智能已成为解决经典化学或化学空间问题的一种可能的方法,这些问题阻碍了药物的发现和开发。随着技术的进步和高性能计算机的发展,计算机辅助药物设计(CADD)中的人工智能算法(如ML到DL)越来越多。人工智能并不是科学家发现和开发药物的新技术;化学家期望准确预测化学活性结构关系也不是新鲜事。例如,Hammett将平衡常数与反应速率联系起来,而Hansch用计算机辅助预测药物化合物的物理化学性质和生物活性。Hansch的成功为以下研究提供了一条途径:(a)化学结构的详细识别和预测,以及药效团和三维结构等性质的表征;(b)假设与预测化合物的化学表征和生物活性相关的复杂数学方程。然而,当今时代科学家的主要目标是通过基于经典化学活动的ML算法来提高药物发现和开发过程的准确性和置信度。这将鼓励化学家识别人工智能技术的潜力,以回答医学化学的两个关键问题,如“下一个化合物应该是什么?”和“合成化合物的过程是什么?”。因此,在过去的二十年里,发展了许多计算药物发现的技术和工具,定量构效关系(QSAR)方法和自由能最小化技术。例如,文献[65]使用机器智能方法(如DT、随机森林(RF)方法、CNN、SVM、LSTM网络和梯度提升机)区分复合细胞活性。在上述模型中,有些模型通过简化的分子输入行输入系统将化合物表示为字符串,直接用作输入数据,而不是任何化学描述符,起到自然语言处理的作用。他们对单个数据集(Z-score=3)和整个数据集(Z-score=5或6)使用了两个不同的截止值。后来,他们合并了九个不同的指标,用于评估模型的精度、准确性、曲线下面积和Cohen的K值。结果表明,梯度提升机具有数据分布均衡的能力。实验结果还表明,经典的ML方法和DL方法可以对复合细胞活性进行分类[65]。类似地,文献[66]使用双QSAR方法预测PAMPA有效渗透率,其中作者使用偏最小二乘(PLS)方案和分层SVM(HSVR)方案开发了经典QSAR模型和基于ML的QSAR模型。作者认为HSVR方案在训练集、测试集和统计分析方面均优于PLS方案。此外,对于新化合物的合成,化学科学家很容易依赖已发表的文献。随着人工智能和分子生物学技术的发展,区分现有药物和新的化学结构变得相对简单。例如,文献[67]应用计算方法筛选中药中的肝毒性成分,而文献[68]使用计算技术展示了系统发育关系、结构-毒性关系和草药成分网络。最近,Zhang等人对一种新的冠状病毒进行了计算分析,作者在其中筛选了对严重急性呼吸综合征(SARS)具有生物活性的不同化合物。随后,对化合物进行ADME和对接分析。结果表明,已有13种中药对新型冠状病毒有较好的抗药性[69]。因此,传统的以化学为导向的药物发现和开发理念与计算药物设计相结合,提供了一个很好的未来研究平台。此外,全世界的系统生物学和化学科学家与计算科学家合作,开发现代ML算法和原理,以促进药物的发现和开发。
5. 利用人工智能和机器学习技术改造传统的计算药物设计
多年来,计算方法在药物设计和发现中发挥了重要作用,改变了药物设计的全过程。然而,许多问题,如时间成本、计算成本和可靠性,仍然与传统的计算方法有关[70,71]。人工智能有可能消除计算药物设计领域的所有瓶颈,也可以增强计算方法在药物开发中的作用。此外,随着基于ML的工具的出现,确定靶蛋白质的三维结构变得相对容易,这是药物发现的关键步骤,因为新药物是基于蛋白质的三维配体结合环境设计的[72,73]。最近,Google的DeepMind (https://github.com/deepmind)设计了一个基于人工智能的工具,该工具根据PDB结构数据进行训练,称为AlphaFold,它可以根据蛋白质的氨基酸序列来预测蛋白质的3D结构[74]。AlphaFold分两步预测蛋白质的3D结构:
(i) 首先,使用CNN将蛋白质的氨基酸序列转换为距离矩阵和扭转角矩阵;(ii)其次,使用梯度优化技术将这两个矩阵转换为蛋白质的三维结构[75]。同样,哈佛医学院的Mohammed AlQuraishi也设计了一个基于数据仓库的工具,以蛋白质的氨基酸序列为输入,生成其三维结构。该模型称为递归几何网络(https://github.com/aqlaboratory/rgn),使用单个神经网络计算连接不同氨基酸的化学键的键角和旋转角,以预测给定蛋白质的三维结构[76]。
此外,量子力学用于确定分子在亚原子水平上的性质,用于估计药物开发过程中蛋白质与配体的相互作用。然而,有时使用传统的计算技术,量子力学的计算可能非常昂贵且要求很高,这会影响其准确性[77]。然而,有了人工智能,量子力学可以变得更加友好和有效。Schtutt等人2019最近开发了一种DL驱动工具,称为SchNOrb(GitHub – atomistic-machine-learning/SchNOrb: Unifying machine learning and quantum chemistry with a deep neural network for molecular wavefunctions),该工具可以准确预测有机分子的分子轨道和波函数。利用这些数据,我们可以确定分子的电子性质、分子周围化学键的排列以及反应位点的位置[78]。因此,SchNOrb可以帮助研究人员设计新药。此外,分子动力学(MD)模拟分析了分子在原子水平上的行为和相互作用[79]。在药物发现中,MD模拟用于评估蛋白质-配体相互作用和结合稳定性。MD模拟的一个主要问题是,它可能非常困难和耗时。AI具有加速MD仿真过程的能力[80]。在这方面,Drew Bennett等人进行了MD模拟,以计算15000个小分子从水转移到环己烷的自由能,从而利用这些自由能和其他一些原子特征来训练3D卷积网络和空间图形CNN。研究人员发现,经过训练的神经网络预测转移自由能的精度与MD模拟计算结果几乎相似[81]。这项研究表明,ML技术可以改进和加快MD模拟。然而,要实现这一点,需要大量的训练数据。
此外,近年来,从头药物设计也利用了人工智能。例如,Q. Bai等人在2020年设计了MolAIcal(MolAICal),这是一种可以在三维蛋白质袋中设计三维药物的工具[82]。MolAICal通过两个组分的作用设计3D药物:(i)第一个组分使用DL和在美国食品和药物管理局(FDA)批准的药物上训练的遗传算法进行从头药物设计,(ii)第二个组分结合分子对接和在ZINC数据库(ZINC)[83]上训练的DL模型。同样,Popova等人2018设计了一种基于深度强化学习的算法,称为ReLeaSE(https:// github.com/isayev/ReLeaSE),用于从头药物设计。ReLeaSE通过集成两个深度神经网络(DNN)(称为生成神经网络和预测神经网络)实现其预期结果,其中生成模型用于生成新化合物,预测模型用于预测化合物的性质[84]。此外,近年来,人工智能也被用于升级合成规划过程,该过程用于确定感兴趣分子的最佳合成路径。最近,Grzybowski等人[85]开发了一个基于DT的程序,称为chematica,用于设计所需分子的新合成路径。类似地,Genheden等人已经实现了AiZynthFinder(https://github.com/MolecularAI/aizynthfin der),这是一种基于Monte Carlo树搜索的反转录合成规划的开源工具,由神经网络调节[86]。同样,Segler等人[87]利用三个不同的神经网络结合Monte Carlo树搜索来发现新的逆合成路线。ICSYNTH(https://www.deepmatter.io/products/ icsynth/)是另一个工具,它可以通过使用通过ML模型生成的一系列化学规则来生成新的化学合成路径[88]。
此外,各种基于文本挖掘的工具也被开发出来,可以帮助传统的药物发现过程。文本挖掘使用自然语言处理(NLP)等方法将各种文献和数据库中的非结构化文本转换为结构化数据,并对其进行适当的分析以获得新的见解。NLP是人工智能的一个分支,它允许计算机通过基于人工智能的算法处理和分析人类语言,如语音和文本。利用这种人工智能驱动的技术,已经开发了各种基于文本挖掘的工具。例如,Jang等人2018利用NLP和主题建模开发了PISTON(http://datab io.gachon.ac.kr/tools/PISTON/),这是一种可以预测药物副作用和药物适应症的工具[89]。同样,DisGeNET(DisGeNET – a database of gene-disease associations)是一个文本挖掘驱动的数据库,包含了大量关于基因疾病和变异疾病关系的信息。DisGeNET中的数据可以分析各种生物过程,如药物不良反应、参与疾病的分子通路、药物对靶点的作用等。此外,STRING(STRING: functional protein association networks)是另一个文本挖掘驱动的数据库,包含各种生物体的蛋白质相互作用的大量信息[91]。此外,STITCH(STITCH: chemical association networks)是另一个文本挖掘驱动的数据库,其中包含蛋白质和化学品/小分子之间相互作用的信息[92]。STICH中的信息也可用于确定药物的结合亲和力和药物-靶点的关联。
6. 人工智能在一、二级药物筛选中的应用
如今,人工智能已经成为一种非常成功且要求很高的技术,因为它节省了时间且具有成本效益[93]。一般来说,细胞分类、细胞分选、计算小分子的性质、借助计算机程序合成有机化合物、设计新化合物、开发分析方法以及预测目标分子的三维结构是一些费时费力的任务,借助人工智能可以减少并加快药物发现的进程[94,95]。药物初筛包括利用人工智能技术通过图像分析对细胞进行分类和分选。许多使用不同算法的ML模型识别图像的精度很高,但在分析大数据时却显得力不从心。要对目标细胞进行分类,首先需要对ML模型进行训练,使其能够识别细胞及其特征,这基本上是通过对比目标细胞的图像来完成的,这将目标细胞与背景分离[96]。提取具有不同纹理特征的图像,如小波纹理特征和Tamura纹理特征,并通过主成分分析(PCA)进一步降维。研究表明,最小二乘支持向量机(LS-SVM)的分类精度最高,为95.34%[97,98]。关于细胞分类,机器需要快速从给定样本中分离出目标细胞类型。有证据表明,图像激活细胞分选(IACS)是最先进的设备,可以测量细胞的光学、电学和机械性能[99][图3]。
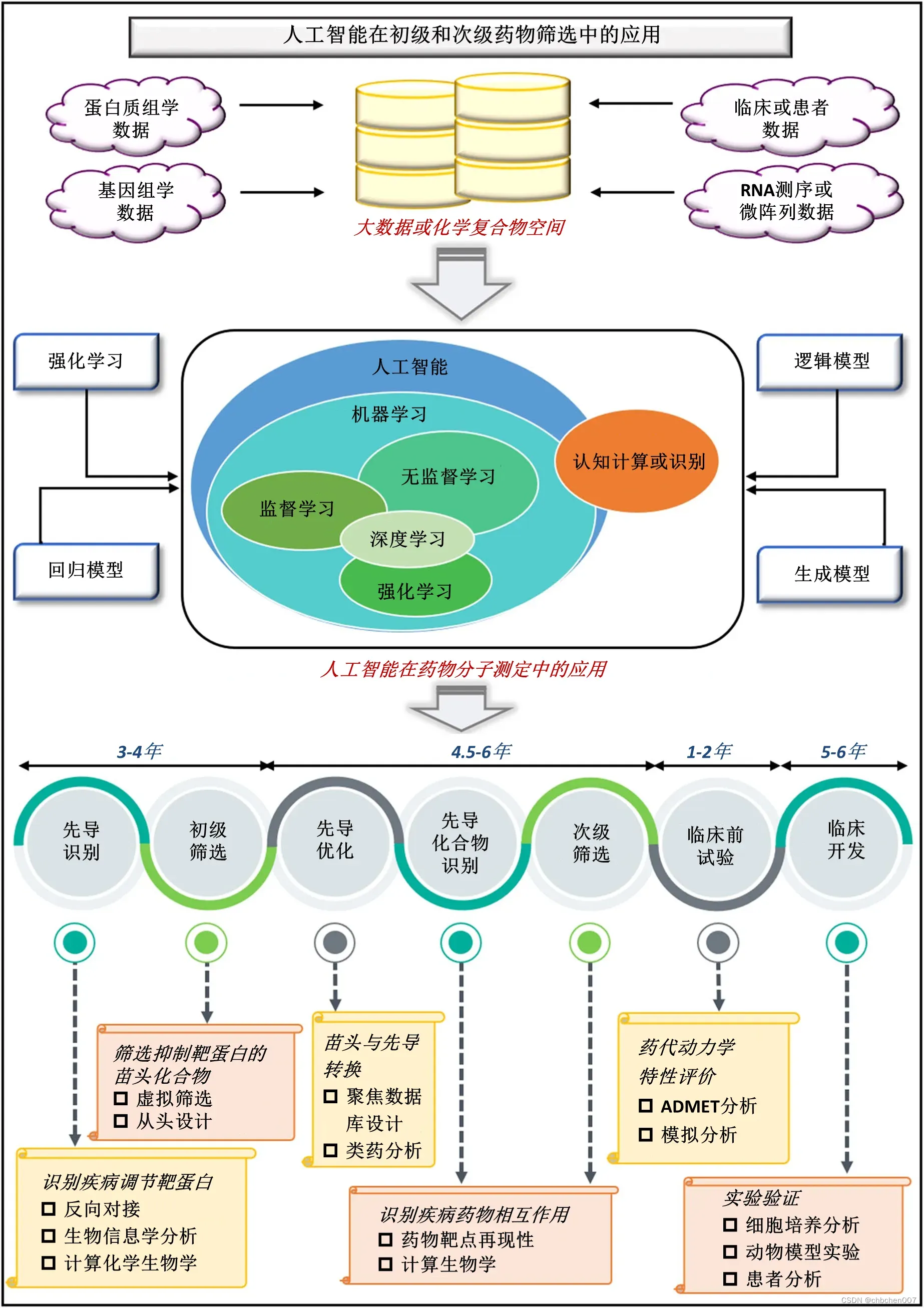 图3 人工智能在一、二级药物筛选中的应用:在药物发现和设计管道中,潜在先导化合物的筛选至关重要,人工智能在识别新的潜在先导化合物方面发挥着重要作用。从组学研究、临床和临床前研究、体内试验和微阵列分析等不同的研究中,化学空间中出现了大约万亿个化学结构。利用强化模型、逻辑模型、回归模型和生成模型等机器学习模型,根据活性位点、结构和靶结合能力筛选出这些化学结构。通过人工智能完成的药物发现过程需要14-18年的时间,相对传统的药物发现过程要短。药物发现过程的第一步是先导识别,通过反向对接、生物信息学分析和计算化学生物学来识别疾病修饰靶蛋白。第二步,对化合物进行初步筛选,筛选出对靶蛋白有抑制作用的潜在先导化合物。这可以通过虚拟筛选和从头设计来实现。药物发现过程的下一步包括通过聚焦库设计、类药物分析、药物-靶点再现性和计算生物学进行先导优化和先导化合物识别。之后,对化合物进行二次筛选,然后进行临床前试验。药物发现过程的最后一步是通过细胞培养分析、动物模型实验和患者分析进行临床开发
图3 人工智能在一、二级药物筛选中的应用:在药物发现和设计管道中,潜在先导化合物的筛选至关重要,人工智能在识别新的潜在先导化合物方面发挥着重要作用。从组学研究、临床和临床前研究、体内试验和微阵列分析等不同的研究中,化学空间中出现了大约万亿个化学结构。利用强化模型、逻辑模型、回归模型和生成模型等机器学习模型,根据活性位点、结构和靶结合能力筛选出这些化学结构。通过人工智能完成的药物发现过程需要14-18年的时间,相对传统的药物发现过程要短。药物发现过程的第一步是先导识别,通过反向对接、生物信息学分析和计算化学生物学来识别疾病修饰靶蛋白。第二步,对化合物进行初步筛选,筛选出对靶蛋白有抑制作用的潜在先导化合物。这可以通过虚拟筛选和从头设计来实现。药物发现过程的下一步包括通过聚焦库设计、类药物分析、药物-靶点再现性和计算生物学进行先导优化和先导化合物识别。之后,对化合物进行二次筛选,然后进行临床前试验。药物发现过程的最后一步是通过细胞培养分析、动物模型实验和患者分析进行临床开发
二级药物筛选包括化合物的物理性质、生物活性和毒性分析。熔点和分配系数是控制化合物生物利用度的一些物理性质,也是设计新化合物的必要条件[100],在设计药物时,可以使用不同的方法进行分子表征,如分子指纹、简化分子输入行输入系统(SMILES)和库仑矩阵[101]。这些数据可用于DNN,DNN包括两个不同的阶段,即生成阶段和预测阶段。虽然这两个阶段通过有监督学习分别进行训练,但当它们联合训练时,偏差可以应用于输出,在输出中,偏差会因特定属性而得到奖励或惩罚。整个过程可用于强化学习[84]。匹配分子对(MMP)已被广泛用于QSAR研究。MMP与候选药物的单一变化相关,这进一步影响化合物的生物活性[102]。与MMP一样,其他ML方法也被使用,如DNN、RF和梯度提升机(GBM)来进行修改。结果表明,DNN的预测能力优于RF和GBM[103]。随着ChEMBL、PubChem和ZINC等公开数据库的增加,我们可以访问数百万种化合物,这些化合物注释了它们的结构、已知靶点和可购买性等信息;MMP+ML可预测口服暴露、内在清除率、ADMET和作用方式等生物活动[98,104,105]。优化化合物的毒性是药物发现中最耗时和最昂贵的任务,也是一个关键参数,因为它为药物开发过程增加了重要价值。
7. 人工智能在药物开发过程中的应用
在药物的发现和开发过程中,最困难和最令人沮丧的步骤是确定存在于浩瀚的化学空间(大约10^60个分子)中的合适的生物活性药物分子。此外,药物发现和开发过程被认为是一个耗时和成本高的过程。最令人恼火的一点是,十分之九的药物分子通常无法通过II期临床试验和其他监管机构的批准[106–108]。上述药物发现和开发的局限性可以通过实现基于AI的工具和技术来解决。AI参与药物开发过程的每个阶段,如小分子设计、药物剂量和相关有效性的确定、生物活性剂的预测、蛋白质-蛋白质相互作用、蛋白质折叠和错误折叠的确定、基于结构和配体的VS、QSAR建模、药物再利用、毒性和生物活性的预测,以及鉴定药物化合物的作用模式,如下所述。
7.1 多肽合成与小分子设计
肽是一种具有生物活性的小分子链,由大约2–50个氨基酸组成,由于其能够穿过细胞屏障并到达所需的靶点,因此越来越多地被用于治疗目的[109]。近年来,研究者利用人工智能技术发现了新的多肽。例如,Yan等人2020开发了Deep-AmPEP30,一种基于DL的短抗微生物肽(AMP)鉴定平台[110]。DeepAmPEP30(AxPEP)是一个CNN驱动的工具,它可以从DNA序列数据预测短AMP。Yan等人利用Deep-AmPEP30从光滑念珠菌(一种存在于胃肠道的真菌病原体)的基因组序列中鉴定出新的AMP。同样,Plisson等人2020将ML算法与异常检测技术相结合,以发现具有非溶血特征的AMP[111]。此外,Kavousi等人开发了IAMPE(http://cbb1.ut. ac.ir/),一个用于鉴定抗菌肽的web服务器,它集成了基于13CNMR的特征和肽的物理化学特征作为ML算法的输入,以识别新的AMP[112]。类似地,Yi等人2019设计了ACP-DL(https://github.com/haichengyi/ACP- DL),一种基于DL的工具,用于发现新的抗癌肽[113]。ACP-DL采用LSTM算法,这是递归神经网络(RNN)的改进版本,用于区分抗癌肽和非抗癌肽。此外,Yu等人[114]提出了DeepACP,一种基于深度递归神经网络的抗癌肽识别模型。同样,Tyagi等人2013开发了一个基于SVM的平台,用于识别新的抗癌肽[115]。此外,Rao等人2020结合图形卷积网络和一种热点编码设计ACP-GCN,用于发现抗癌肽[116]。此外,Grisoni等人使用四个反向传播神经网络的集成来识别新的抗癌肽。同样,Wu等人[117]提出了PTPD,一种基于CNN和word2vec的工具,用于发现用于治疗的新肽。
此外,小分子是分子量非常低的分子,像肽一样,小分子也被用于基于人工智能的工具的治疗目的。例如,Zhavoronkov等人[118]设计了生成性张量强化学习(GENTRL),这是一种基于生成性强化学习的小分子从头设计工具。在GENTRL(GitHub – insilicomedicine/GENTRL: Generative Tensorial Reinforcement Learning (GENTRL) model)的帮助下,Zhavoronkov等人发现了一种新的酶抑制剂,DDR1激酶[118]。同样,McCloskey等人[119]将DNA编码的小分子库(DEL)数据与Graph CNN和RF等ML模型相结合,发现了新的类药物小分子。类似地,Xing等人[120]整合了XGBoost、SVM和DNN,以寻找类风湿性关节炎靶点的小分子。
7.2 药物剂量和给药有效性的鉴定
给患者服用不适当剂量的任何药物都可能导致不良和致命的副作用;因此,为治疗目的确定安全的药物剂量至关重要。多年来,确定一种药物的最佳剂量一直是一个挑战,这种药物可以在毒副作用最小的情况下达到预期的疗效[121]。随着人工智能的出现,许多研究者正借助ML和DL算法来确定合适的药物剂量。例如,Shen等人[122]开发了一个基于AI的平台,称为AI-PRS,以确定通过抗逆转录病毒治疗用于艾滋病毒治疗的最佳剂量和药物组合。AI-PRS是一种神经网络驱动的方法,通过抛物线反应曲线(PRS)将药物组合和剂量与疗效联系起来。在他们的研究中,Shen等人对10名HIV患者联合使用替诺福韦、伊法韦仑和拉米夫定,并在适当的时候使用PRS方法,他们发现替诺福韦的剂量可以减少起始剂量的33%,而不会导致病毒复发。因此,使用AI-PRS也可以找到其他疾病的最佳用药剂量。
此外,Pantuck等人[123]开发了CURATE.AI,以确定适当的药物剂量,它使用患者的个人数据并将其转换为CURATE.AI配置文件,以确定最佳剂量。这项研究是在癌症药物恩扎鲁胺和研究药物ZEN-3694联合应用于转移性去势抵抗前列腺癌患者的情况下进行的。使用CURATE.AI,随着时间的推移,他们发现比ZEN-3694的起始剂量低50%,可以达到预期的效果并阻止癌症生长。
此外,Julkunen等人[124]设计了comboFM(https:// github.com/aalto-ics-kepaco/comboFM),这是一种新的ML驱动工具,可在临床前研究(如癌细胞系)中确定合适的药物组合和剂量。comboFM通过使用因子分解机(https://github.com/geffy/ tffm)确定合适的药物组合和剂量,这是一个用于高维数据分析的ML框架。在他们的研究中,Julkunen等人利用comboFM鉴定了一种新的抗癌药物克唑替尼和硼替佐米的组合,在淋巴瘤细胞系中显示出良好的疗效。类似地,Sharabiani等人使用ML方法确定抗凝药物华法林的最佳初始剂量。他们使用相关向量机根据患者的剂量需求对不同患者进行分类,然后使用回归模型预测患者的合适剂量[125]。同样,Nemati等人[126]开发了一个深度强化学习模型,该模型在重症监护II数据库(MIMIC II)中进行多参数智能监测,以找到另一种抗凝药物肝素的理想剂量。同样地,Tang等人[127]使用ML技术,如ANN、贝叶斯加性回归树、增强回归树、多元自适应回归样条来确定免疫抑制药物他克莫司的最佳剂量。此外,Hu等人[128]采用分类回归树、多层感知器网络、k近邻等技术进行ML分析,以确定心脏药物地高辛的安全初始剂量。此外,Imai等人[129]开发了一个DT模型,以找到抗生素万古霉素的安全起始剂量。
7.3 生物活性物质的预测与药物释放的监测
类药物的设计和监测是一个繁琐和耗时的过程。最近,多个在线工具被开发来分析药物释放和检查所选择的生物活性化合物作为载体的责任。随后使用基准数据集验证计算分析。对于这样的评价,基于化学特征的药效团最适合。这些模型构建大型3D数据集,这些数据集是通过电子实验或内部化合物收集开发的[130]。为了研究基于配体的化学特性,已经使用CATALYST程序(www.accelerys.com)建立了各种成功的实验,并且一组研究人员使用VS实验[131]成功地预测了11种β-羟基类固醇脱氢酶1型抑制剂。
确定生物活性配体是筛选针对特定靶点的有效药物的关键步骤。现在,研究人员正在利用人工智能确定可用于与疾病相关的特定靶点的生物活性化合物。例如,Wu等人综合DL和RF方法设计WDL-RF(https://zhanglab. ccmb.med.umich.edu/WDL-RF/),以确定G蛋白偶联受体(GPCR)靶向配体的生物活性。同样,Cichonska等人[132]开发了pairwiseMKL(KEPACO · GitHub),一种基于多核学习的方法,用于确定化合物[133]的生物活性。为了测试他们的模型的有效性,他们用以预测化合物的抗癌效力。此外,Mustapha等人[134]开发了一个Xgboost模型,以确定生物活性化学分子。此外,Merget等人[135]创建了DNN、RF等机器学习模型,以确定280多种不同激酶的生物活性。此外,Arshadi等人[136]设计了DeepMalaria,一种基于DL的模型,用于识别具有恶性疟原虫抑制活性的化合物。同样,Sugaya等人[137]创建了配体效率驱动的支持向量回归模型,以确定各种化合物的生物活性。此外,Afolabi等人[138]使用MLD药物数据报告(MDDR)存储库中的数据,并将其应用于增强算法组合,以识别新的生物活性化合物。此外,Petinrin等人[139]使用多数投票技术和不同机器学习模型的集成来确定生物活性分子。
此外,药物不良反应(ADR)是由给药引起的意外、有害、致命的副作用。ADR是药物开发中的一个主要挑战,在药物开发的初期识别可能的ADR以使药物开发过程更加稳健和有效已成为必要。最近,研究人员使用人工智能来确定不同药物在上市前可能出现的不良反应。例如,Dey等人[140]使用了基于DL的模型,该模型可以预测与药物相关的不良反应,甚至可以识别导致这些不良反应的化学亚结构。此外,Liu等人[141]综合了药物的化学、生物学和表型特性,通过机器学习分析预测与之相关的ADR。同样,Jamal等人[142]结合生物、化学和表型特性,通过机器学习分析预测与药物相关的神经系统不良反应。作者还利用他们的模型找出了与目前阿尔茨海默病药物相关的不良反应。此外,Xue等人[143]将生物医学网络拓扑与DL算法相结合,以预测药物ADR相关性。此外,Raja等人[144]使用机器学习分析预测ADR,ADR是药物相互作用的结果。他们进一步使用他们的模型来预测与皮肤疾病药物相关的ADR。除了筛选有效的生物活性剂外,另一个关键领域是药物的相似性及其相互作用后释放。最近,开发了一个可自由访问、用户友好的图形界面SwissADME(http://www.swissadme.ch),以评估药物的相容性及其药代动力学作用[145]。Higuchi、Hixson–Crowell、Ritger–Peppas–Kormeyers、Brazel–Peppas、Baker–Lonsdale、Hopfenberg、Weibull和Peppas–Sahlin等数学模型也已应用于药物发现,最常见的做法之一是计算选定或筛选的生物活性分子的载药量。
7.4 蛋白质折叠和蛋白质相互作用的预测
分析蛋白质相互作用(PPI)对于有效的药物开发和发现至关重要。大多数蛋白质注释方法使用的序列同源性范围有限。高通量的蛋白质相互作用数据,随着数据量的不断增加,正成为新的生物学发现的基础。生物信息学面临的一个巨大挑战是对这些数据进行管理、分析和建模。因此,计算模型的发展,在一个地方同时预测多个输入[146]。虽然计算方法被认为比蛋白质-蛋白质非相互作用(PPNI)更具信息性,但也暗示了研究PPI和蛋白质-蛋白质非相互作用(PPNI)的计算方法。PPI预测可分为直接PPI、间接功能关联的直接PPI和信号转导途径的PPI[147]。利用机器学习和统计学习方法,如K近邻、朴素贝叶斯、支持向量机、神经网络、DT和RF等,对PPI中的障碍进行预测。贝叶斯网络(BN)基本上是利用基因共表达、基因本体(GO)等生物过程相似性来预测PPI的。使用BN的数据集集成产生精确和准确的PPI网络,说明全面的酵母相互作用组[148]。另一组还使用BN结合酵母的数据集来研究PPI[149]。一种新的层次模型PCA集成极限学习机(PCA-EELM)仅利用蛋白质序列信息预测蛋白质-蛋白质相互作用,是一种有效的工具,具有输出准确、持续时间短等优点。此外,通过一种称为DNN蛋白质相互作用预测(DeepPPI)的新方法(http://ailab.ahu.edu.cn:8087/DeepPPI/index.html)[151]提高了DNS PPI预测效率。在哺乳动物细胞中,信号转导主要由非结构基序和球状蛋白结合域(PBD)之间的PPI控制。为了跨多个蛋白质家族预测这些PBD,开发了定制的ML工具,称为分层统计力学建模(HSMM)[152]。基于ML、结构域亲和力和频率表的蛋白质-蛋白质相互作用预测,一种称为PPI_SVM的新工具于2011年开发,可在(http://code.google.com/p/cmater-bioinfo/)[153]免费访问。由于已解决的复杂结构的数量不断增加,已经开发了一种多体线程方法MULTIPROPERTOR。在该方法中,将已知模板结构的蛋白质重新思考,并建立其与其他蛋白质的相互作用、界面能和Z评分[154]。基于结构的线程逻辑回归工具Struct2Net(http://struct2net.csail.mit.edu)用于评估交互概率,是除同源建模之外的第一个基于结构的PPI预测工具[155]。基于基因簇的方法计算同一基因簇编码的查询蛋白同源序列的共现概率。这种方法也称为结构域/基因共现。如果两个蛋白质的基因在基因组中并不接近,那么这种方法就不能可靠地预测这两个基因之间的相互作用[156,157]。
7.5 基于结构和配体的虚拟筛选
在药物设计和药物发现中,VS是CADD的关键方法之一。VS是指识别与药物靶点结合的小化合物。VS是从化合物库中筛选出有前景的治疗性化合物的有效方法[158]。因此,它成为高通量筛选的重要工具,同时也带来了成本高、准确率低的问题。一般来说,有两种重要的VS类型,即基于结构的VS(SBV)和基于配体的VS(LBV)[159,160]。LBV依赖于活性配体和非活性配体的化学结构和经验数据,它利用活性配体的化学和物理化学相似性,从具有高生物活性的化合物库中预测其他活性配体。然而,LBV不依赖于靶蛋白的三维结构,因此,该方法在缺少靶结构或信息且获得的结构精度较低的情况下实现[161]。另一方面,SBVS已经在这样的情况下实现,其中蛋白质或靶的三维结构信息已经通过体外或体内实验或通过计算建模阐明[162,163]。通常,该方法用于预测活性配体或其相关靶点之间的相互作用,以及预测参与药物-靶点结合的氨基酸残基。与LBVS相比,SBVS具有较高的准确性和精密度。然而,SBVS与越来越多的致病蛋白及其复杂构象有关[164]。要将ML用于VS,应该有一个由已知活性和非活性化合物组成的过滤训练集。这些训练数据用于使用监督学习技术训练模型。然后对训练后的模型进行验证,如果模型足够精确,则将该模型用于新的数据集,以筛选针对靶具有所需活性的化合物[165]。之后,入围的化合物可以进行ADMET分析,然后进行各种生物测定,然后进入临床试验。因此,ML具有加速VS的能力,使其更加健壮,甚至可以减少VS中的误报。对接是SBVS中应用的主要原则,其中开发了几种基于AI和ML的评分算法,如NNScore、CScore、SVR Score和ID-Score[166]。类似地,已经构建了诸如RFs、SVM、CNN和浅层神经网络等ML和DL方法来预测SBV中的蛋白质-配体亲和力。此外,基于人工智能的算法已被开发用于SBV[167]中的分子动力学模拟分析。另一方面,LBVS由几个步骤组成,每个步骤都提出了新的基于AIA和ML的算法,以加快过程并提高可靠性。例如,已经构建了一些基于ML和DL的算法来准备有用的诱饵集,例如高斯混合模型(GMM)、隔离林和人工神经网络(ANN)。
此外,已为LBV[168,172]构建了PARASHIFT、HEX、USR和ShaPE算法等ML模型。目前,随着人工智能算法在医疗和制药行业的兴起,针对LBV和SBV开发了不同的工具和模型。例如,已经为SBV开发了MTiOpenScreen(http://bioserv.rpbs. univ-paris-diderot.fr/services/MTiOpenScreen/)[170]、FlexX‐Scan[171]、CompScore(http://bioquimio.udla.edu.ec/compscore/)[172]、PlayMolecule BindScope(PlayMolecule.org)[173]、GeauxDock(http://www.brylinski.org/ geauxdock)[174]、EasyVS(http://biosig.unimelb.edu.au/ easyvs)[175]、DEKOIS 2.0[176]、PL-PatchSurfer2(http://www.kiharalab.org/plps2/)[177]、SPOT-ligand 2(http:// sparks-lab.org/)[178]、Gypsum -DL(https://durrantlab.pitt. edu/gypsum-dl/)[179]和ENRI[180]等工具。此外,越来越多的证据验证了AI在SBV中起关键作用的假设,如识别非肽半胱氨酸-半胱氨酸趋化因子受体5受体激动剂[181],筛选β2肾上腺素能受体的部分激动剂[182],识别含溴代多巴胺的蛋白4抑制剂[183],天然产物样信号转导子和转录激活因子3二聚化抑制剂[184]的发现,VHL和缺氧诱导因子1-α抑制剂[185]的预测,以及Kelch样ECH相关蛋白核因子红系2相关因子2(Keap-Nrf2)小分子抑制剂[186]的预测。同样,Liu等人2017年发现了低毒的O-GlcNAc转移酶抑制剂,而Dou等人[187]通过SBV[188]发现了新的糖原合成酶激酶3β(GSK-3β)抑制剂。通过SBVS对肿瘤和白血病进行了不同的研究,如发现了治疗急性髓系白血病的新型GSK-3β[189],在非小细胞肺癌中鉴定了新型蛋白精氨酸甲基转移酶5抑制剂[190],鉴定了治疗肾癌的血管内皮生长因子受体2强效化合物[191],乳腺癌多靶点抑制剂[192]的鉴定及Mdm2-p53抑制剂的发现[193]。近年来,新型冠状病毒在世界范围内成为一个巨大的问题,因此,SBVS也为化学和生物科学家提供了一个很好的机会来识别针对致病靶点的新型药物化合物。例如,Gahlawat等人2020确定沙奎那韦、紫草次酸和11m_32045235是抗SARS-Cov-2主要蛋白酶的有希望的治疗性化合物,而Selvaraj等人2020证明TCM 57025、TCM 3495、TCM 5376、TCM 20111和TCM 31007是与N7-MTase底物结合位点相互作用的治疗性化合物[194,195]。同样的趋势,Cruz等人2018得出结论,ZINC91881108是针对RIPK2的有效化合物,而Simoben等人2018证明了八种新型N-(2,5-二氧吡咯烷-3-基)-羟肟酸二烷基酯衍生物作为smHDAC8抑制剂,针对smHDAC8,IC50值范围为4.4至20.3 µM [196,197][图4]。
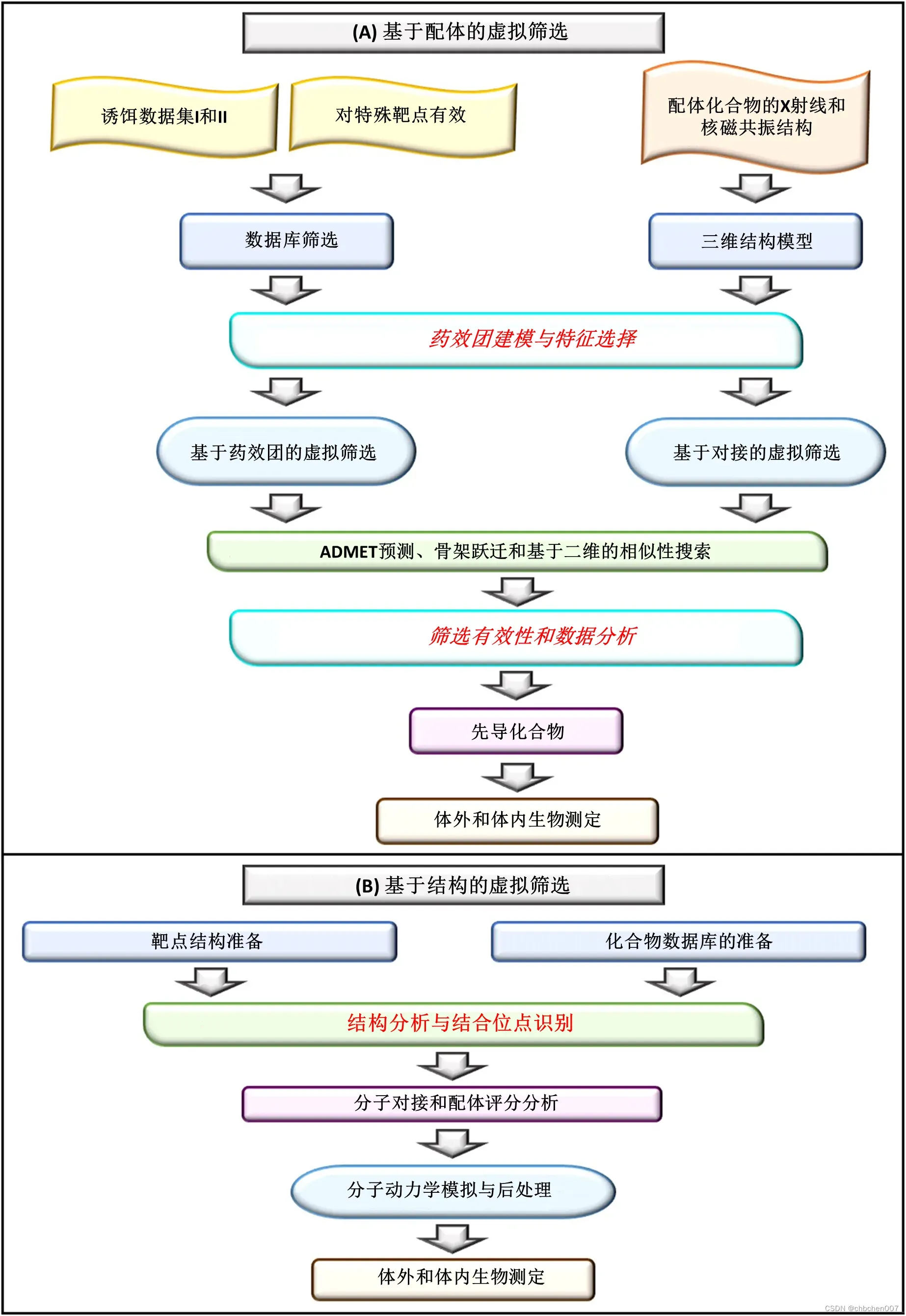 图4 (A)基于配体的虚拟筛选:在药物设计和发现过程中,基于配体的虚拟筛选是最关键的步骤,包括如图所示的不同步骤。初始步骤包括数据库筛选和三维结构模型通过特定靶标的活性位点和复合物的X射线结构进行预测。随后,对具有选定特征的选定化合物进行药效团建模,然后对化合物进行基于药效团和对接的虚拟筛选。筛选出的化合物具有不同的毒性和理化性质,以供进一步分析。最后,对先导化合物进行体外和体内生物测定以进行验证。(B)基于结构的虚拟筛选:它是应用于药物发现过程中的另一种虚拟筛选,其中靶结构准备和化合物库准备是初始步骤。然后,进行结构分析和结合位点预测,然后将化合物与选定的靶点进行分子对接。随后,进行了分子动力学模拟研究,以在生物信息学中验证筛选出的化合物,然后通过生物测定进行实验验证
图4 (A)基于配体的虚拟筛选:在药物设计和发现过程中,基于配体的虚拟筛选是最关键的步骤,包括如图所示的不同步骤。初始步骤包括数据库筛选和三维结构模型通过特定靶标的活性位点和复合物的X射线结构进行预测。随后,对具有选定特征的选定化合物进行药效团建模,然后对化合物进行基于药效团和对接的虚拟筛选。筛选出的化合物具有不同的毒性和理化性质,以供进一步分析。最后,对先导化合物进行体外和体内生物测定以进行验证。(B)基于结构的虚拟筛选:它是应用于药物发现过程中的另一种虚拟筛选,其中靶结构准备和化合物库准备是初始步骤。然后,进行结构分析和结合位点预测,然后将化合物与选定的靶点进行分子对接。随后,进行了分子动力学模拟研究,以在生物信息学中验证筛选出的化合物,然后通过生物测定进行实验验证
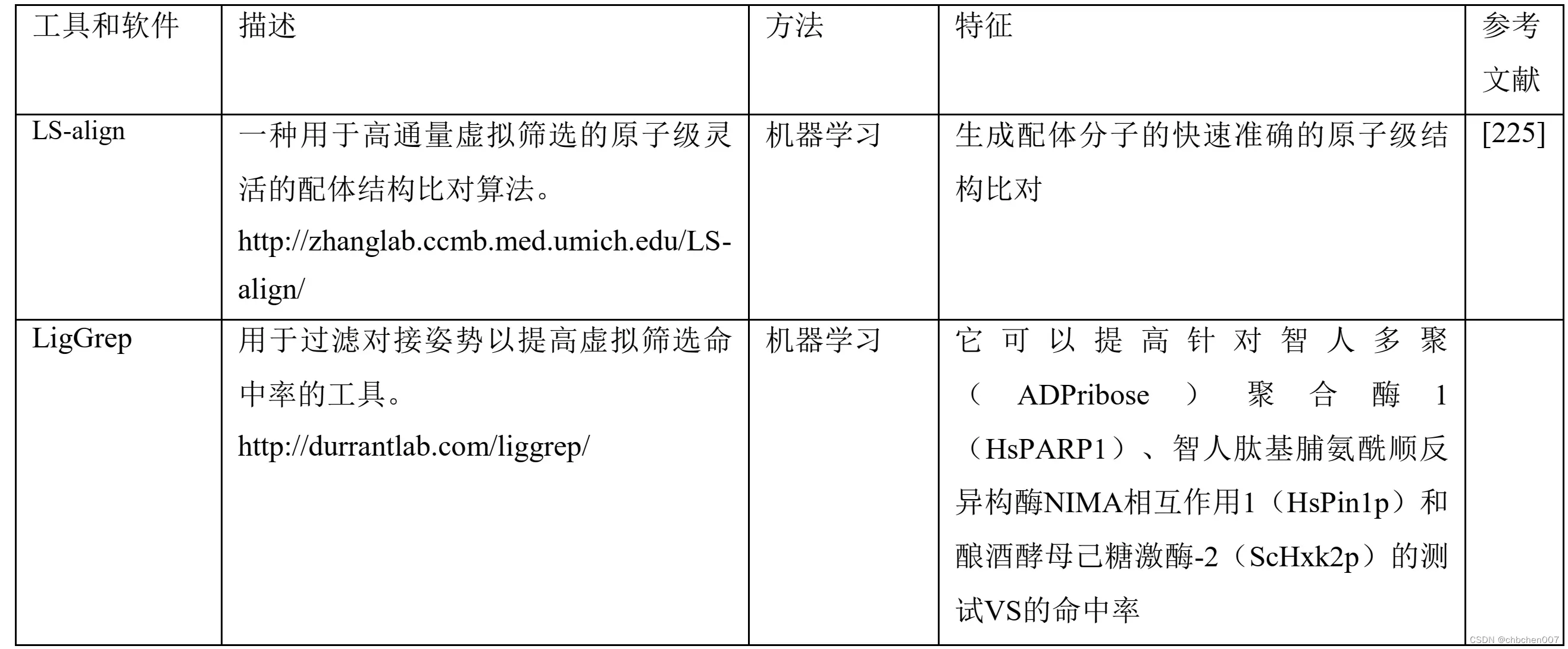
此外,针对LBV开发了不同的算法和工具,如SwissSimilarity(http://www.swiss similarity.ch/)[189]、METADOCK[199]、开源平台[200]、HybridSim VS(http://www.rcidm.org/Hybri dSim-VS/)[201]、PKRank[202]、PyGOLD(http://www. agkoch.de/)[203]、BRUSELAS(http://bio-hpc.eu/softw are/Bruselas)[204]、RADER(http://rcidm.org/rader/)[205]、QEX[206]、IVS2vec(https://github.com/haiping1010/ IVS2Vec)[207]、AutoDock Bias(http://autodockbias.wordp ress.com/)[208]、Ligity[209]、D3Similarity(https://www. d3pharma.com/D3Targets-2019-nCoV/D3Similarity/index. php)[210]和GCAC (http://ccbb.jnu.ac.in/gcac)[211]。新证据表明,人工智能算法在LBV中的潜在应用,如识别极光激酶A抑制剂[212]、G-四链体靶向化学型[213]、PI3Kα抑制剂[214]、靶向登革热病毒非结构蛋白3螺旋酶[215]、潜在的选择性组蛋白去乙酰化酶8抑制剂[216],以及新型对羟基苯丙酮酸双加氧酶抑制剂[217]。除上述研究外,许多文献证实了AI在LBV中的可能实施,例如识别HIV侵入抑制剂和有效的DNA甲基转移酶抑制剂[218,219]。与SBV一样,LBV在识别针对新型人类冠状病毒的潜在治疗化合物方面也起着关键作用。例如,Amin等人2020展示了一些内部分子作为木瓜蛋白酶样蛋白酶抑制剂的分子对接研究,而Hofmarcher等人2020至DNN从库中鉴定了30000种化合物作为CoV-2抑制剂[220,221]。同样,Choudhary等人2020确定SARS-CoV-2细胞进入抑制剂,而Ferraz等人2020确定贝达喹啉、格列本脲和咪康唑为抗冠状病毒的潜在治疗化合物[222,223]。Xiao等人2018针对六个抗癌靶点,针对化合物库的VS开发了基于配体的大数据DNN模型。该研究整合了0.5 M的化合物,并通过十倍交叉验证对所开发的模型进行了评估[224]。随着化合物库规模的不断扩大,寻找潜在的苗头化合物变得越来越困难,这就像大海捞针一样。因此,SBV和LBV在最小化潜在治疗性化合物识别的复杂性方面发挥着巨大的作用。此外,SBV和LBV中基于AI的模型使其更简单,具有高准确度和高精确度。表1讨论了在LBV和SBV中实现的基于AI和DL的不同web工具和算法。
表1 包括机器学习(ML)和深度学习原理在内的人工智能(AI)算法在基于结构和配体的虚拟筛选中的应用
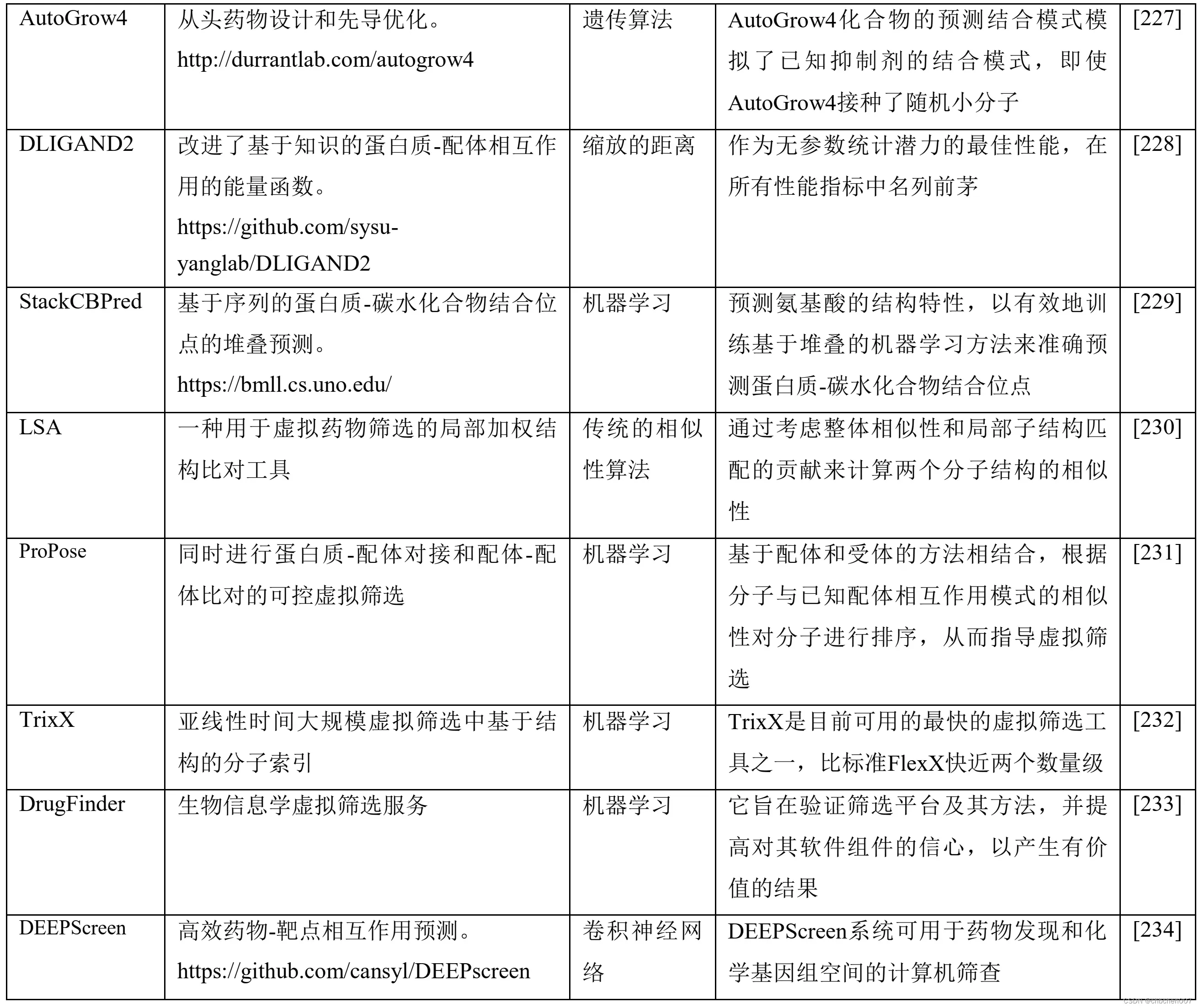
7.6 QSAR模型与药物再利用
在药物的设计和发现中,研究药物的化学结构、理化性质与生物活性之间的关系至关重要。因此,QSAR建模是一种计算方法,通过它可以建立化学结构和生物活性之间的定量数学模型。建立一个数学模型的主要优点是从分子数据库中识别出各种各样的化学结构,这些结构可以作为针对疾病靶点的治疗性化合物。一旦选定最有希望的化合物,就要进行实验室合成和体外或体内试验。QSAR模型大致分为回归模型和分类模型两类。高斯过程(Gaussian Process,GP)是一种建立QSAR回归模型的方法,是一种稳健而有效的QSAR建模方法。GP方法可以处理大量的描述符,并识别出关键的描述符。近年来,已有两种分类模型被证明是使用GP这一固有的GP分类方法,另一种是GP回归技术与probit分析相结合的方法[235,236]。此外,该方法适用于非线性关系的建模,并且不需要主观地确定模型参数[237]。近年来,神经网络、DL、SVM等ML算法的发展和应用日益广泛,为QSAR建模提供了一条新的途径。已经开发了几种基于web的QSAR建模工具和算法,如VEGA平台(https://www.vega-qsar.eu/)[238]、QSAR-Co(https://github.com/meta-QSAR/simple-tree)[239]、FL-QSAR(https://github.com/bm2-lab/FL-QSAR)[240]、Meta-QSAR(https://github.com/meta-QSAR/simple-tree)(https://github.com/meta- QSAR/drug-target-descriptors)[241]、DPubChem(www.cbrc.kaust.edu.sa/ dpubchem) [242], Transformer-CNN(GitHub – bigchem/transformer-cnn: Transformer CNN for QSAR/QSPR modelling)[243]、Cloud 3D-QSAR (http://chemyang. ccnu.edu.cn/ccb/server/cloud3dQSAR/)[244]、Transformer-CNN(GitHub – bigchem/transformer-cnn: Transformer CNN for QSAR/QSPR modelling)[243]、Cloud 3D-QSAR(http://chemyang.ccnu.edu.cn/ccb/server/cloud3dQSAR/)[244]、MoDeSuS和ChemException(https:// github.com/Abdulk084/Chemception)[245]。Karpov等人2020开发了一种新的基于ANN的QSAR建模算法,称为transformer-CNN。该方法利用SMILES增强进行训练和干扰。类似地,Wang等人2020通过集成分子结构生成、排列和分子相互作用场的特征,开发了基于web的QSAR建模工具。Jin等通过Cloud 3D-QSAR发现了一种高效、选择性的单胺氧化酶B(MAO-B)抑制剂。在本研究中,作者认为(S)-1-(4-((3-氟苄基)氧基)苄基)氮杂环丁烷-2-甲酰胺(C3)比番红花酰胺(safinamide)更有效和选择性地抑制MOB。此外,体内分析显示,化合物C3可抑制脑MAO-B活性并挽救1-甲基-4-苯基-1,2,3,6-四氢吡啶(MPTP)诱导的多巴胺能神经元丢失[246]。基于同样的趋势,Bennett等人2020通过化学感受,结合MD模拟和DL[81]预测了小分子转移自由能。此外,QSAR协同工具还应用于不同的研究中,如I类磷脂酰肌醇3-激酶酶亚型抑制的多靶点化学计量学模型的开发,ERK抑制剂作为抗癌药物的筛选,K562细胞功能抑制剂的预测,以及酚类化合物抗真菌特性的预测[247-250]。同样,Kim和Cho 2018开发了一种称为PyQSAR(https://github.com/crong-k/pyqsar_tutorial)的新算法,用于使用ML和Jupyter笔记本的快速QSAR建模平台。PyQSAR是一个独立的python包,它将所有QSAR建模过程组合在一个工作台中[251]。A. S. Geoffrey等人2020使用PyQSAR进行了两项不同的研究,例如识别新型冠状病毒的有效候选药物和开发槲皮素及其肿瘤坏死因子α抑制活性的QSAR[252,253]。此外,Zuvela等人开发了基于人工神经网络的QSAR模型,用于预测黄酮类化合物的抗氧化活性。在这项研究中,作者整合了六种方法,如PaD、PaD2、权重、逐步、扰动和轮廓,用于解释和阐明基于ANN的模型,该模型计算植物当量的抗氧化性能。结果表明,基于神经网络的算法可以消除由于描述分子结构的量子力学参数解释不当而产生的困难[254]。同时,Ding等人2020生成了一个基于web的工具,称为VISAR(GitHub – Svvord/visar),用于通过DNN QSAR方法剖析化学特征[255]。越来越多的证据表明,QSAR模型在药物设计和发现过程中的应用,如与肝脂肪变性中AOP分子起始事件相关的ToxCast分析建模[256],针对二肽基肽酶8和二肽基肽酶9酶的二肽基肽酶4抑制剂的开发[257],QSAR模型在HIV-1蛋白酶抑制剂[258]结构域分析及靶向HIV/HCV合并感染[259]研究中的应用。ML模型的一个公认问题是SAR模型生成中生物测定数据缺失值的数据插补。基本上有三种主要类型的缺失值:(i)完全随机缺失(MCAR),当变量中缺失值的概率对于所有样本都相同时,就会出现这种情况;(ii)随机缺失(MAR),这意味着变量中随机缺失值的概率仅取决于其他预测值中的可用信息;(iii)非随机缺失(MNAR),这意味着缺失值的概率不是随机的,并且取决于未记录的信息和现有信息预测缺失值[260]。有几种方法可以处理缺失值,如使用零、均值、中值或模式公共值进行插补,使用随机选择的值进行插补,使用模型进行插补或使用深度学习库(Datawig)进行插补。每个数据集都缺少需要明智地处理的值,以便构建健壮的模型[261]。此外,还应消除数据的复杂性,并对数据进行整理,以提高生成的模型的准确性和精确度。此外,最初的QSAR模型用于预测小分子(如分子量(mw)小于1500 m.w的分子)的毒性和代谢。然而,2000年代初应用的QSAR技术存在一些限制,如准确性和可靠性[262]。随着QSAR在药物发现和设计过程(如VS、先导优化和靶点识别)中的日益广泛应用,医学科学家和生物学家不断努力开发更可靠和可信赖的方法[263]。基于AI/ML算法的QSAR模型具有消除早期方法约束的潜力。基于AI/ML的QSAR模型,即基于全息图的QSAR(HQSAR)、基于基团的QSAR(G-QSAR)和基于集成的QSAR模型,使药物发现过程加速了数倍[264,265]。此外,除了经典的Hansch和Free-Wilson方法外,QSAR在过去几年中随着新的细化方法、描述符计算的新方法、系统验证测试的实施以及受体结构信息的参与而逐渐发展。类似地,除了经典的先导优化外,QSAR已应用于药物发现和设计的不同新兴领域,如肽QSAR、混合物毒性QSAR、纳米粒子QSAR、离子液体QSAR、化妆品QSAR、植物化学QSAR和材料信息学[266][图5]。
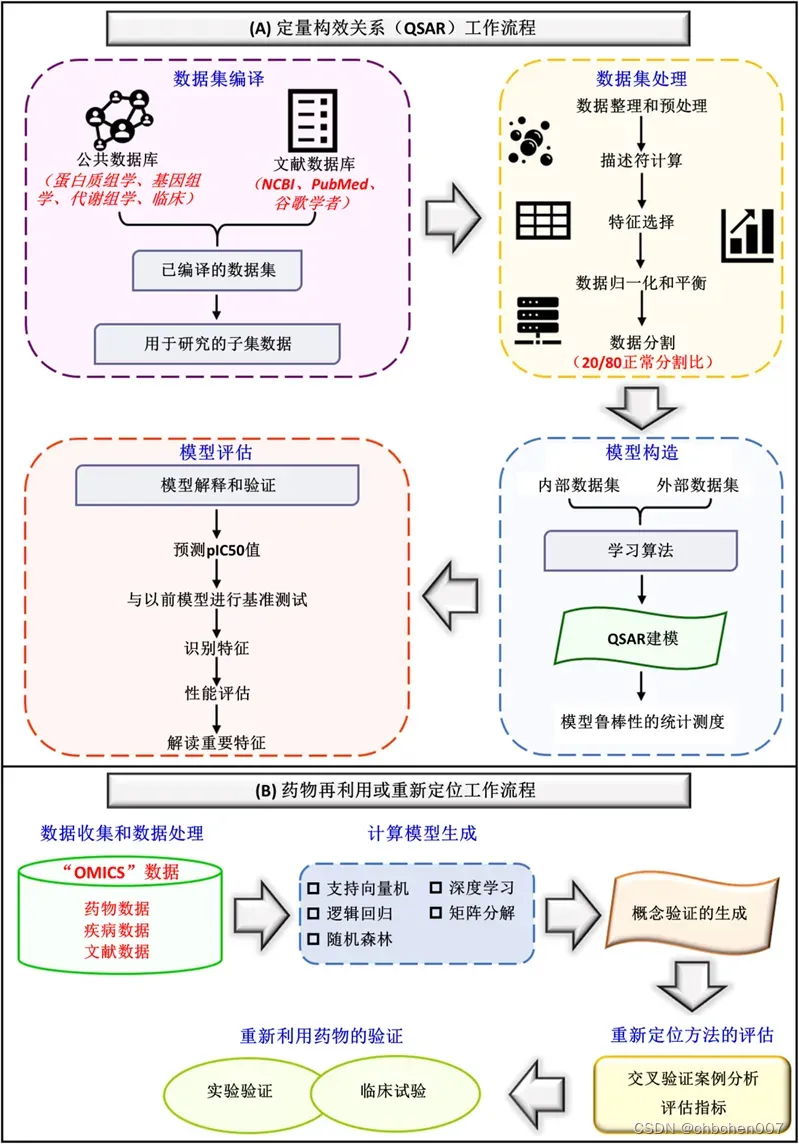
图5 (A)定量结构-活性关系工作流:初始步骤包括数据集编译,其中来自公共数据库和文献数据库的数据被累积和编译,进一步划分为不同的子集进行调查。然后,进行数据集处理,其中数据预处理和整理,然后计算分子描述符。在描述计算之后,执行数据集处理、数据归一化以及将数据拆分为不同的数据集。第三步进行模型构建,积累内部数据和外部数据等数据集,应用学习算法进行QSAR建模。最后,通过统计计算来衡量模型的鲁棒性。定量结构-活性关系的最后一步是模型评估,通过与以前的基准模型进行比较、识别特征、性能评估和解释基本特征来评估模型。(B)药物再利用或重新定位工作流程:第一步是数据收集和数据预处理,然后是计算模型生成。生成的模型包括支持向量机、逻辑回归、随机森林、深度学习和矩阵分解。然后,从文献来源生成概念证明。随后,通过交叉验证、案例分析和评估度量来评估重新定位模型。最后,通过临床试验、体外研究和体内研究对再利用药物进行验证
除了QSAR建模外,人工智能算法还应用于药物再利用或药物再定位方法中。在药物设计和发现中,药物重新定位是指对已开发的药物进行研究,并将其重新定位到其他疾病状态。由于多个靶点参与多种疾病的可能性,重新定位药物可能是成功的[267-269]。另一方面,基因组学、蛋白质组学和体内外药理学研究的大数据集的出现为药物的重新定位提供了一个很好的途径。近年来,基于人工智能的药物发现工具和算法的出现为今后的研究提供了平台。ML算法以新的系统生物学方法取代了基于化学相似性和分子对接的传统方法,可以评价药物作用[270-273]。因此,最近开发了不同的基于AI的算法和基于web的工具,如DrugNet(http://genome2.ugr.es/ drugnet/)[274]、DRIMC(https://github.com/linwang1982/ DRIMC)[275]、DPDR-CPI(http://cpi.bio-x.cn/dpdr/)[276]、PHARMGKB(PharmGKB)[277]、Promiscular 2.0(http://bioinformatics.charite.de/promiscuous2)[278]和DRRS(http://bioinformatics.csu.edu. cn/resources/softs/DrugRepositioning/DRRS/index.html)[279]。此外,Yella和Jegga等人2020年利用称为MGATRx的多视图图注意方法构建了药物重新定位模型[280],而Yan等人2019年基于称为BiRWDDA的多相似性融合方法构建了药物重新定位的新算法[281]。此外,Fahimian等人于2020年构建了一种称为RepCOOL的新算法,用于识别有希望用于II期乳腺癌的再利用药物。结果表明,阿霉素、紫杉醇、曲妥珠单抗和他莫昔芬是治疗II期乳腺癌的潜在药物[282]。同样,Li等人2020构建了一个基于宿主的药物再利用的计算框架,用于针对RNA病毒的广谱抗病毒药物。在这项研究中,作者调查了2352种已批准的药物和1062种天然化合物对不同的病毒病原体,并得出结论,重新利用的药物对寨卡病毒和冠状病毒有效[283]。此外,Wu等人2020应用ML模型,即结构谱预测模型和生物谱预测模型,预测抗纤维化候选药物。结果表明,训练集的受试者工作特性曲线下面积分别为0.879和0.972,而测试集的受试者工作特性曲线下面积分别为0.814和0.874。结果表明,天然产物具有抗纤维化特性,可作为潜在的抗纤维化药物靶点。最近,COVID-19作为一种全球流行病出现,全球各地的研究人员开始寻找有希望的治疗药物。在这方面,基于人工智能的药物重新定位起着至关重要的作用。例如,基于网络的药物再利用确定了16种潜在的抗HCoV再利用药物,而Hooshmand等人2020基于多模式DL方法确定了12种有希望的COVID-19药物靶点[285,286]。近年来,神经网络、DL模型和药物定位管道的发展在很大程度上得到了提高。例如,基于药物相似性网络融合的SNF-CVAE识别出治疗阿尔茨海默病(AD)和幼年类风湿性关节炎的有希望的治疗药物,而基于神经网络算法和整合长-短期记忆的DTI-RCNN预测药物-靶点相互作用[287,288]。PhenoPredict和SDTNBI是另外两种基于ML的算法,分别用于识别精神分裂症疾病全表型药物重新定位和预测药物-靶相互作用[289,290]。Zang等人2019开发了一个基于DL的模型,称为deepDR(GitHub – ChengF-Lab/deepDR),用于预测生物信息学药物的重新定位。在本研究中,作者整合了药物-疾病、药物-副作用、药物-靶点等10种不同类型的生物网络和7种药物-药物网络。结果表明,deepDR预测利培酮和阿立哌唑等药物可用于治疗阿尔茨海默病(AD),而哌甲酯和培高利特可用于治疗帕金森病(PD)[291]。同样,Chen等人2020构建了一种基于人工智能的新算法iDrug(https://github.com/Case-esaC/iDrug),用于通过跨网络嵌入整合药物重新定位和药物靶点预测。iDrug的效率和有效性允许用户了解药物-靶点-疾病机制的新临床见解[292]。研究表明,通过基于人工智能的算法进行药物再利用可以在癌症中实现。例如,Li等人2020使用DL整合转录组学数据和化学结构信息,并确定吡莫唑是治疗非小细胞肺癌的有希望的候选药物[293]。类似地,Kuenzi等人2020使用人类癌细胞的DL模型预测药物反应和协同作用。结果表明,预测组合可提高无进展生存率,反应预测可将ER阳性乳腺癌患者的临床结果分层[294]。另一个AI在药物再利用中的应用来自Wang等人2020年进行的研究,该研究使用二部图卷积网络进行计算机药物再利用。作者通过DL和异构信息融合构建了一个称为BiFusion(https://github.com/zcwang0702/BiFusion)的模型。结果表明,BiFusion在药物再利用方面的性能优于多个基线[295]。上述例子总结了基于人工智能的算法在药物再利用中的潜在作用。此外,随着技术的进步,化学科学家、生物科学家和计算科学家都在寻找提高人工智能模型准确性和精确度的方法。此外,QSAR和药物重定位的药物发现方法都是不完整的,没有分子对接的参与,分子对接用于分析靶分子和配体分子之间的相互作用。最初,在21世纪初,分子对接作为一种独立的工具被开发出来,用于确定作为靶分子和配体分子的两个分子之间的相互作用。然而,随着人工智能技术的出现,分子对接的适用性发生了变化。现在,分子对接正被用于与MD模拟和基于AI的工具结合,用于药物发现的不同领域,如VS、靶点识别、多重药理学和药物再利用[296]。MD模拟和人工智能算法的实现可以提高分子对接的效率和精度。此外,多年来,分子对接的局限性也得到了解决。例如,在药物设计中,分子对接只适用于晶体结构可用的生物靶点,因为许多靶点的结构是不可用的。因此,一种类似同源建模的技术被开发出来以克服这一障碍[297]。此外,PDB中的晶体结构数据呈指数增长,增强了分子对接在药物发现中的适用性。表2讨论了已在计算机QSAR和药物重新定位中实现的工具和算法。
表2 包括机器学习(ML)和深度学习原理在内的人工智能(AI)算法在药物设计和发现过程中的应用
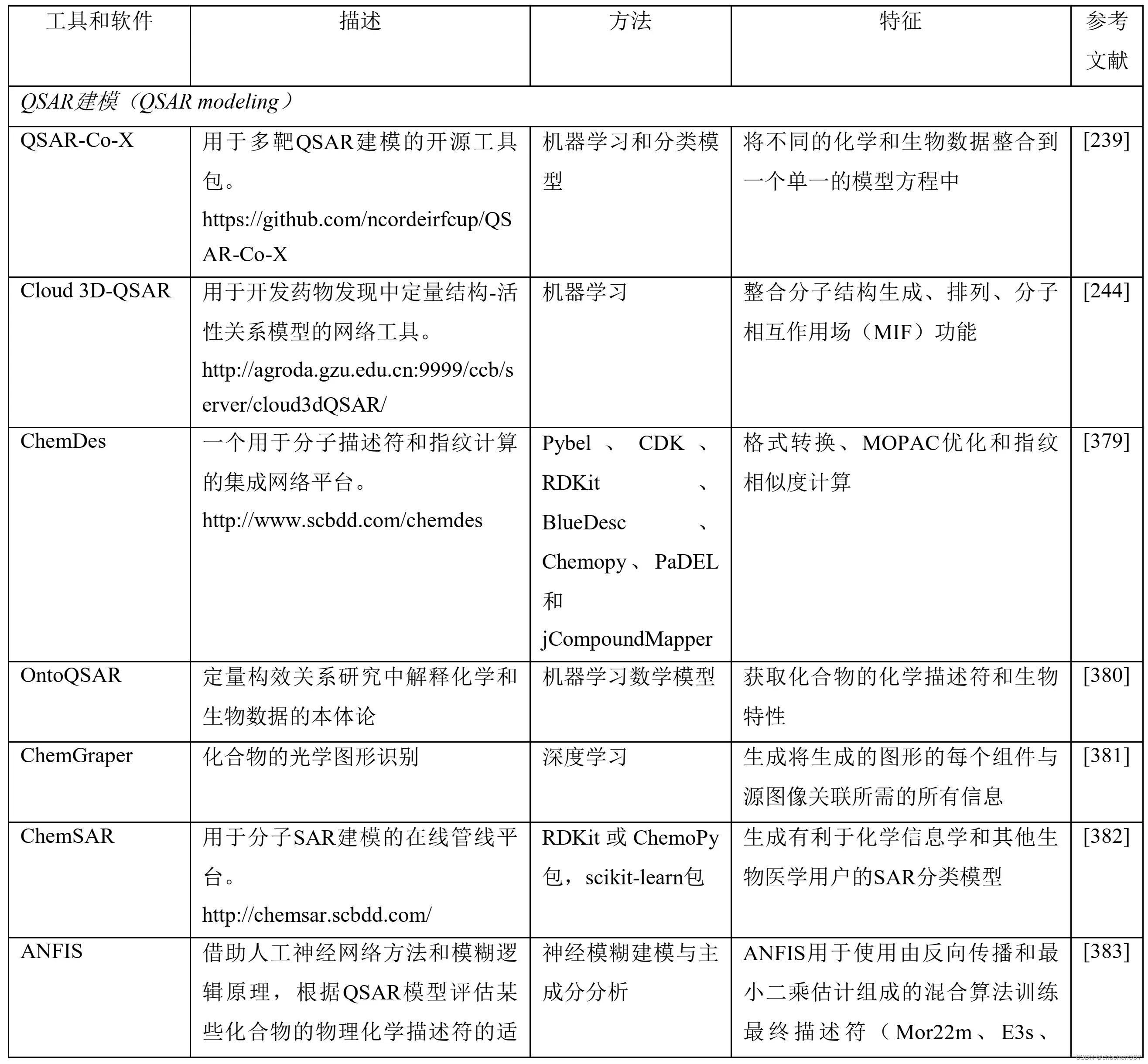
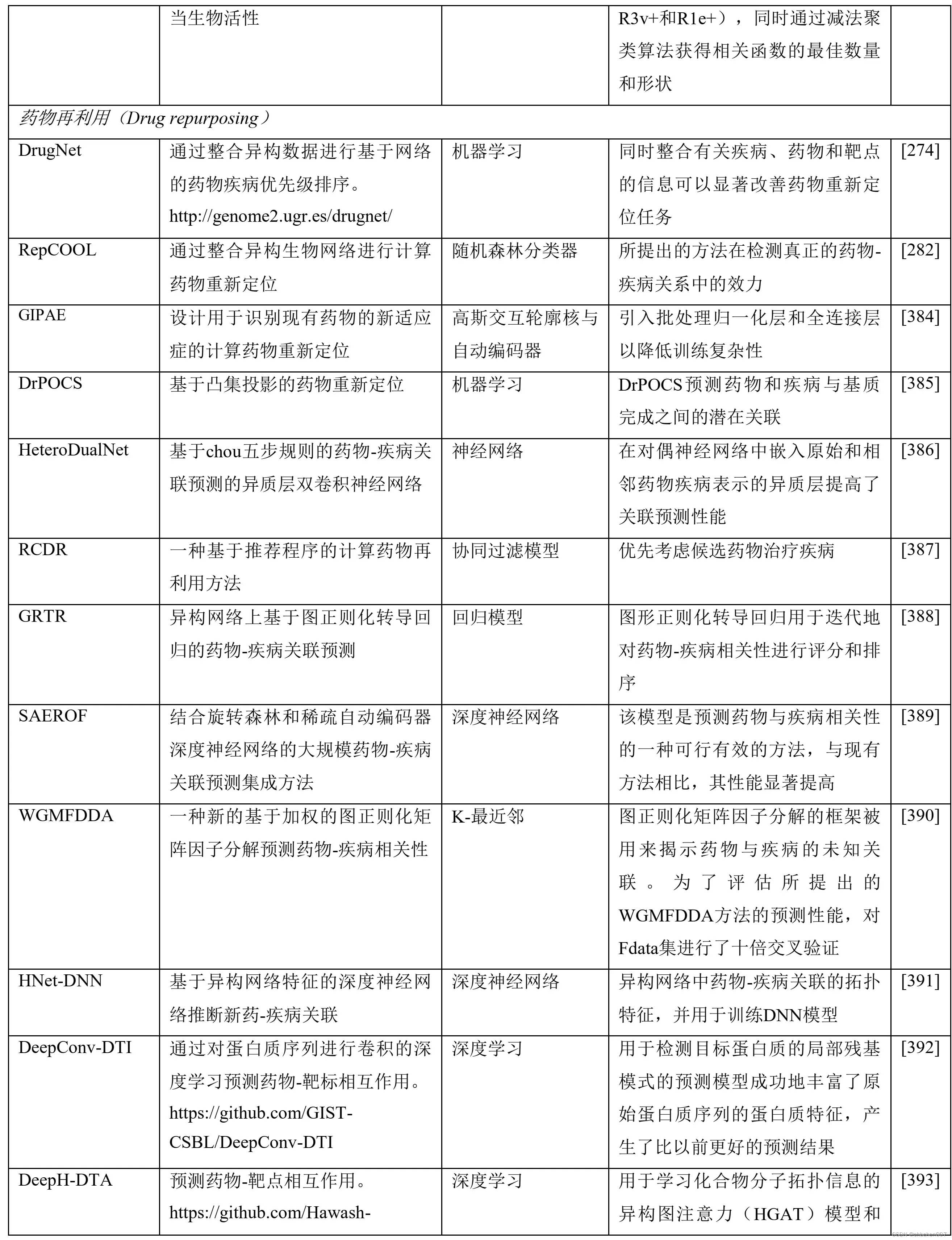
7.7 理化性质及生物活性预测
每个化合物都与溶解度、分配系数、电离度、渗透系数等理化性质有关,这可能会影响化合物的药动学性质和药物-靶点结合效率。因此,在设计新的药物分子时,必须考虑化合物的物理化学性质[100,298]。为此,开发了不同的人工智能工具来预测化合物的物理化学性质。用于预测化合物的生物物理和生化性质的基于AI的工具包括分子指纹、SMILES格式、库仑矩阵和势能测量,它们用于DNN训练阶段[299,300]。最近,Zhang等人开发了一个QSAR模型来预测从美国环境保护署(EPA)提取的六种不同理化性质的环境因子。类似地,Lusci等人2013构建了基于神经网络的模型来预测分子性质。在该研究中,分子是用无向循环图来描述的,而以前的预测物理化学性质的方法是用有向无环图来描述的[301]。随后,构建了6种基于人工智能的化合物人体肠道吸收预测算法。构造的方法有支持向量机、k近邻、概率神经网络、人工神经网络、偏最小二乘法和线性判别模型。在上述模型中,SVM具有较高的准确率91.54%[302]。2016年,Zang等人开发了一个基于ML的模型,用于预测环境化学品的物理化学性质,如辛醇-水分配系数、水溶性、沸点、熔点、蒸汽压和生物浓缩因子[303]。此外,还开发了不同的基于AI的工具,如ALOGPS 2.1(On-line Lipophilicity/Aqueous Solubility Calculation Software)[304]、ASNN(Associative Neural Network)[305]、E-BABEL(http://www.vcclab.org/lab/babel/)[304]、PCLIENT(http://www.vcclab.org/lab/pclient/)[304]、E-DRAGON(http://www.vcclab.org/lab/edragon/)[304]、ChemSpider(http://www.chemspider.com/)[306]、SPARC(http://sparc.chem.uga.edu/sparc/)[307]和OSIRIS property explorer(https:// www.organic-chemistry.org/prog/peo/)[308]。2020年开展了双苯并咪唑类抗癌药物的设计、合成及ADMET预测研究。在同一研究中,作者通过Lipinski的五定律计算了化合物的分子性质,并预测了合成化合物的预ADMET性质[309]。此外,Puratchikody等人2016年在其研究中使用ORISIS property explorer来预测用于安全、有效炎症治疗的酪氨酸衍生物的定量结构毒性。结果表明,在55个有效分子中,只有19个被认为是有效的环氧化酶-2抑制剂。在相似的实验条件下,建立了基于RF和DNN的人体肠道吸收模型。因此,从实例可以看出,基于人工智能的方法通过预测药物的理化性质在药物发现和开发中具有重要作用。
此外,药物分子的治疗活性取决于其与受体或靶点的结合效率,因此不能表现出与药物靶点结合亲和力的化学分子将不被视为治疗剂。因此,预测化学分子与治疗靶点的结合亲和力对于药物的发现和开发至关重要[311]。人工智能算法的最新进展增强了结合亲和力预测的过程,它利用药物及其相关靶点的相似性特征。已经开发了一些基于web的工具,如ChemMapper和相似集成方法(SEA)。此外,已经构建了基于ML和DL的药物-靶点亲和力识别模型,例如KronRLS、SimBoost、DeepDTA和PADME[312]。KronRLS通过预测药物与其靶点之间的相似度来计算基于ML算法的药物-靶点结合亲和力。KronRLS在预测药物-靶点结合亲和力时同时考虑了基于特征和基于相似性的相互作用[313]。DeepDTA(https://github.com/hkmztrk/DeepDTA)[314]和PADME[315]等DL方法预测药物-靶点结合亲和力,这取决于蛋白质的三维结构。Beck等人2020年进行了一项研究,通过DeepDTA[316]预测商业上可获得的抗病毒药物作为一种潜在的治疗剂,用于对抗新型冠状病毒(SARS-CoV-2)。类似地,Lee和Kim 2019基于使用PADME的大规模药物诱导转录组数据,预测了DNN的药物-靶相互作用[317]。构建了一个同时使用RNN和CNN预测药物-靶点结合亲和力的DL模型,称为DeepAffinity (GitHub – Shen-Lab/DeepAffinity: Protein-compound affinity prediction through unified RNN-CNN)[318]。Jiang等人2019年利用DeepAffinity提出了一种新的蛋白质描述符,用于识别药物-靶点相互作用,而Born等人2020年利用Deep Affinity确定了SARS-CoV-2的候选抗病毒药物[319,320]。以上数据验证了ML和DL算法在药物设计中对药物分子的理化性质和生物活性的重要性。然而,从研究的角度来看,这些算法的有效性和准确性仍然是一个显著的缺点。因此,需要进行广泛的研究,通过精确和广泛的数据输入来最大限度地提高基于人工智能的算法的准确性和精度。在表2中,我们总结了基于人工智能算法的理化和生物活性预测工具和数据库,包括DL、神经网络、SVM等。
7.8 化合物的作用方式和毒性预测
药物毒性是指由于化合物的作用或代谢方式,化学分子对生物体或生物体任何部分的不利影响。人工智能的扩展范围有可能预测药物分子的非靶向和靶向效应,以及化合物合成前的体内安全性分析,已吸引了与药物开发过程相关的科学家。人工智能的参与减少了药物开发时间、成本、损耗率和人力资源。为此,开发了不同的基于web的工具,如LimTox(http://limtox.bioinfo.cnio.es/)[321]、pkCSM(pkCSM)[322]、admetSAR(http://lmmd.ecust.edu.cn/admetsar2/)[323]和Toxtree(Toxtree – Toxtree – Toxic Hazard Estimation by decision tree approach)[324]。Srivastava等人2020使用admetSAR评估Withania somnifera作为抗COVID-19治疗化合物的毒性,而Uygun等人2021结合pkCSM来鉴定吡唑并[1,5-a]吡嗪-4(5H)-酮衍生物对肺腺癌细胞系的治疗作用和毒理学特性[325,326]。基于AI的方法的进步导致了不同毒性预测软件和基于web的工具的开发,如Tox21(Tox21)[327]、SEA(SEA Search Server)[328]、eToxPred(https://www.brylinski.org/etoxpred-0)[329]和TargeTox(https://github.com/ artem-lysenko/TargeTox)[330]。Tox21评估了12707种环境化合物和药物的毒性,而SEA预测了656种上市药物对73个非预期目标的毒性。TargeTox基于靶点-药物生物网络预测毒性风险。2016年,Huang等人通过建模Tox21预测了1万多种化合物的体内毒性曲线和机制表征,而同年,Zhou等人利用改进的分子SEA预测了癌症相关蛋白质[331,332]。此外,Gupta和Rana 2019年采用eToExpred预测雄激素受体小分子的毒性。作者在10273种药物中纳入了1444种小分子特征,其中461种为活性药物,9812种为非活性药物[333]。
DeepTox(http://bioinf.jku.at/research/DeepTox/tox21.html)[334]和PrOCTOR(https://github.com/kgayvert/PrOCTOR)[335]分别用于预测新化合物的毒性和预测临床试验中的毒性概率。例如,Robledo Cadena等人2020使用PrOCTOR预测了非甾体抗炎药对顺铂、紫杉醇和阿霉素对宫颈癌细胞的疗效的影响,而Gilvarie等人2020确定了2576个小分子的新适应症,这些小分子与PD和2型糖尿病的16种不同药物特征相结合[336,337]。同样,Simm等人2018使用DeepTox分析并重新利用高通量成像分析数据,以预测针对替代生物途径和过程的不同化合物的生物活性[338]。此外,DeepTox还用于开发多个ML和DL算法,用于预测药物化合物的毒性特性和化学特性,如SMILES2Vec(预测化学特性)[339]、Chemiception(基于DNN的化学特性预测)[245]、DeepSynergy(使用DL预测抗癌药物协同作用)[340],和deepAOT(复方急性经口毒性预测)[341]。然而,可以通过使用大型和精细化的数据集来提高DeepTox和PrOCTOR的准确性和精确度,这可以通过制药行业的参与来实现。最近,其他基于ML的工具,如SPIDER[341]和阅读-跨结构-活性关系(read-Cross structure–activity relationships,RASAR)[342]也被开发出来,它们能够分别分析β-拉帕酮靶点和连接未知化合物的分子结构和毒性特性。
Zhang等人[344]基于五种ML算法结合MACCS或FP4指纹图谱建立了不同的药物肝毒性预测模型。结果表明,最佳模型对外部验证数据集[344]的准确率为75%。基于相关向量机(RVM)、正则化RF、C5.0树、极值梯度提升(XGBoost)、AdaBoost、SVM 提升(SVMBoost)、RVM 提升(RVMBoost)等ML方法构建了毒性评价算法。构建的模型用于评估大鼠口服急性毒性、呼吸毒性和尿路毒性[345-348]。近年来,深度学习算法的应用为化合物的分子表征带来了新的方法,使得DL方法适合于预测化合物的毒性。此外,DL算法用于毒性预测的潜力取决于数据集的质量和数量。总之,要使基于人工智能的毒性预测算法更可靠,还需要进一步的研究。然而,目前基于ML的预测因子仍然不适合替代生物系统,但它们足以将药物化学原理扩展到正确的方向,从而减少合成周期。此外,表2中讨论了基于AI的毒性预测算法和工具的详细描述。
7.9 分子通路识别与多重药理学
AI和ML算法在药物发现和开发中的重要成果之一是预测和估计疾病网络或药物-药物相互作用或药物-靶点关系的整体拓扑和动力学[349]。这种方法为识别特定疾病的新分子治疗靶点提供了广阔的途径。文本挖掘驱动的数据库DisGeNET、STITCH、STRING分别被广泛用于确定基因-疾病关联、药物-靶点关联和分子通路。例如,Gu等人2020使用相似性集成方法识别197种最常用中药的靶点。后来,DisGeNET数据库被用来将这些药物靶点与不同的疾病联系起来,从而将草药与可用于治疗的疾病联系起来[350]。此外,Chen等人2019年使用STITCH数据库寻找食管癌候选药物的靶点[351]。同样,Taha等人2020年使用STITCH数据库寻找家养南天竹(Nandina domestica)活性成分的靶点,家养南天竹是一种用于治疗各种肿瘤的植物。后来使用STRING数据库,借助cytoscape工具[352]构建复合靶通路。
在药物化学中,多重药理学是指在与疾病相关的药物-靶点生物网络中设计一个能够与多个靶点相互作用的单一药物分子。它最适合设计一种有前途的治疗剂,用于治疗更复杂的疾病,如癌症、神经退行性疾病(NDD)、糖尿病、心力衰竭和其他许多疾病[353-355]。基于ML的方法具有强大的挖掘能力和数据分析能力,有可能通过关联分子网络分析犯罪。此外,ML模型通过生成具有所需多重药理学特征的化合物来协助多靶配体的合理设计,因为ML模型生成具有不同化学和拓扑特征的大量化学结构。因此,发现多靶配体的概率增加。此外,ML模型有助于识别存在不同结合口袋的多靶配体。人工智能在药物发现和开发方面的最新进展导致了用于多重药理学预测的基于web的工具和独立软件包的产生,如多重药理学浏览器(PPB)(Reymond Research Group)[356]、TarPred(http://www.dddc.ac.cn/ tarpred/)[140]、基于自组织映射的药物等效关系预测(SPiDER)(http://modlabcadd.ethz.ch/software/spider )[357]、Targethunter(https://www.cbligand.org/TargetHunter3D/)[358]、PharmMapper(http://lilab-ecust.cn/pharmmapper/)[359]、ChemMapper(http://lilab.ecust.edu.cn/chemmapper/)[360],和Swiss Target Prediction(SwissTargetPrediction)(http://www.swisstargetprediction.ch/ )[361]。Poirier等人2018使用PPB进行了一项实验,以确定溶血磷脂酸酰基转移酶β作为纳摩尔血管生成的治疗靶点,而Ozhathil等人2018使用PPB[362,363]确定了阳离子通道瞬时受体电位阳离子通道亚家族M成员4的强效和选择性小分子抑制剂。此外,Vleet Van等人2018实施了TarPred工具,用于筛选改进的非目标责任预测策略和方法,而同年,Ratnawati等人使用反向传播算法预测SMILES代码中的活性化合物[364,365]。在上述基于web的工具中,PharmMapper和ChemMapper经常用于当前的研究。如黄芪、黄连对糖尿病[366]的协同作用机制研究、当归补血汤[367]的补血机制研究、白花蛇舌草多机制预测等。对于结直肠癌[368],使用PharmMapper。类似地,在癌症中鉴定人铜转运阻断剂[369],通过化学蛋白质相互作用在AD中鉴定多靶点配体[370],预测苦参注射液抗肝癌的抗癌机制[371],以及发现基于蝶呤-7(8H)酮的抗表皮生长因子受体激酶T790M/L858R突变体的治疗化合物[372],使用ChemMapper进行。人工智能算法用于多药预测的一个主要限制是数据不足或数据集的可靠性。因此,应进行量子化学计算,提供微调的数据集,从而提高预测模型的准确性。
此外,药物开发中的人工智能为通过基因组学信息、生化特征和靶点说明确定治疗人类疾病的分子通路或分子靶点打开了大门[373]。“OpenTargets”( Home – Open Targets)[374],一个基于ML的免费工具,用于优先考虑潜在的治疗药物靶点,准确率超过71%。最近,Nabirotchkin等人鉴定了常用抗COVID-19药物的未折叠蛋白反应和自噬相关途径,而Lopez-Cortes等人鉴定了结直肠癌中的等位基因频率[375,376]。此外,Isac-Lopez等人[377]进行的GWAS研究预测了多个风险位点,并强调了纤维化和血管病变途径。结果表明,27个独立的全基因组相关信号和13个新的危险位点与系统性硬化相关。Martin等人研究染色质相互作用以预测风湿性疾病中的新基因靶点。在同一研究中,作者得出结论,454个高置信度基因与风湿性疾病相关,其中48个是药物靶点,11个是现有靶点。最后,他们证明367种药物适合重新定位[378]。
8. 人工智能在新药设计中的应用
设计受体三维结构以产生新分子的迭代过程称为从头药物设计,旨在产生新的动力学。然而,从头药物设计并没有在药物披露中看到无限的应用。此外,由于人工智能领域的进步,该领域最近出现了一些复苏[421,422]。VS已成为药物改进措施中的一个巨大工具,因为它在大量混合物中进行了有利可图的计算机检查,进一步扩大了潜在药物线索的产量。作为AI的一个子集,ML是一种协调药物先导物的VS的技术,通常包括收集一组过滤过的化合物,其中包含已知的活性和非活性化合物,以训练模型[423,424]。在建立模型之后,将对其进行测试,如果足够准确,将其用于之前未知的数据库,以识别新药物。在本节中,我们将讨论人工智能如何被证明是使用从头技术进行药物设计的福音。
在一项研究中,研究人员利用惰性空间描述来准备一个依赖于药物相似性定量估计(QED)药物相似性评分和制造可用性评分合成可及性评分(SAS)的模型[425]。在另一个分布中,这种变分自动编码器的呈现与对抗自动编码器的呈现形成对比[426]。该不良布置的自动编码器包括传递新颖复合结构的生成模型。第二个判别拮抗模型用于区分真实粒子和生成粒子,而生成模型则试图欺骗判别模型[427]。从本质上讲,对抗式自动编码器比变分式自动编码器在生成模式下产生了更多的实体结构。在与电子模型的混合中,可以得到预期针对多巴胺受体类型2动态的新结构。研究利用生成不良处置组织(GAN)提出具有假定抗癌特性的混合物[428]。
RNN同样被有效地用于从头药物设计。由于SMILES字符串在一组字母中编码物质结构,RNN被用来生成复合结构。据观察,RNNs有潜力利用SMILES串进行药物设计[429]。类似的方法同样有效地用于开发新的肽结构[430]。神经网络学习被有效地应用于将所创建的混合料倾向于所需的特性[431]。同样,转移学习也被用作另一个系统,以创造具有理想自然行为的新型合成结构。在接下来的步骤中,组织准备使用大量的准备集[432,433]熟悉SMILES语法。在随后的步骤中,用具有理想运动的混合物进行制备。此外,额外的训练时间足以达到新的组合阶段,进入由动态原子组成的复合空间。根据这样一种方法,五个原子被结合起来,四个粒子对抗原子、化学受体的计划行动可以得到确认[434]。一些独特的设计已经被提出,创造了合法的,重要的新结构。新的合成已经研究了这些策略,与性质传播所创建的分子或原子类似的广泛使用的训练集。该策略的主要应用是充分的,5个原子中有4个表示理想作用[435]。人工智能和多目标优化是连接化学和生物两个阶段的有效途径。开发了用于基于SMILES的自动化从头设计的基于RNN的新型多目标对,以找到物理化学性质与其受约束的生物目标之间的最佳匹配。结果表明,人工智能和多目标优化可以捕捉化学和生物方面的潜在联系,从而为可定制的设计策略提供易于使用的选项,这对潜在客户开发和潜在客户优化都特别有效[436]。
ML模型如SVM、RF、DNN等已被用于药物发现,用于分析从对接到VS[437]的药物应用。最近,药物再利用已成为一种创新的方法,以尽量缩短药物开发时间,通常涉及数据挖掘和人工智能[438]。一个小组提出了一个问答人工系统(QAAI),该系统能够利用Google语义AI通用编码器来计算嵌入红脑JSON数据库中的句子。这项研究验证了脂氧合酶抑制剂药物齐留通作为NRF2通路调节剂在体外的预测,在减少巨噬细胞M1表型和活性氧产生方面具有潜在的应用前景。这种新方法已被证明对NDD的重新定位有效[439]。随着系统药理学和多靶点药理学的快速发展,开发合理设计多靶点药物的方法迫在眉睫。第一个全新的多靶点药物配置程序LigBuilder V3(http://www.pkumdl.cn/ligbuilder3/)被设计用于设计不同受体的配体、一个受体的多个偶联位点或一个受体的不同配置。LigBuilder V3再次用于多靶点药物计划和增强,特别是用于具有不同配体结合位点的蛋白质的紧凑配体[440]。从头药物设计积极寻求使用一套化学规则,快速有效地识别具有所需生物学特性的结构新的化学型。此外,基于片段的从头设计工具已成功应用于非共价抑制剂的发现。在此,设计了一种称为Cov_FB3D的新方案,该方案涉及通过识别靶蛋白[441]的共价结合位点中的活性片段来进行潜在新型共价抑制剂的电子组装。
9. 人工智能在药物生产和临床试验设计中的作用
在制药工业中,计算方法的使用是相当成熟的。然而,人工智能的引入为开发能够改进和优化药物发现的新方法提供了更大的范围[442]。这不仅鼓励了科学界,也导致了制药行业和人工智能公司之间日益增长的伙伴关系[443]。一项研究表明,2013年21143种药物的总体成功率接近5.2%,而2005年则低于11.2%。因此,人工智能的使用主要与减少损耗和成本的需要有关[444]。一种新药上市通常需要12年时间,成本高达30亿美元[445]。此外,在现有1060个类药物分子的情况下,寻找新药是一项艰巨的任务[446]。目前药物研发面临的挑战涉及药物的毒性、副作用、选择合适的靶点、合适的剂量甚至知识产权等[447]。制药行业在药物获得批准之前,通常不共享药物的药代动力学和药效学测量。除此之外,用于训练AI模型的药物发现数据非常少[448]。需要一个能够规范和管理临床前和临床药理学数据的团体来加速人工智能在这一领域的进展。人工智能的最新进展在许多方面影响了临床药理学,如文献检索和处理、与在线预测ML模型的交互作用、在许多国家制定政策鼓励医疗保健的ML方法以及获得药物相关信息的预测分析[449,450]。
当候选药物成功通过所有临床前试验后,将其应用于临床试验患者,包括三个阶段:第1阶段,少数人进行药物安全性试验;第2阶段,对少数受特定疾病影响的受试者进行药物疗效试验;第3阶段,对大量患者进行疗效研究,并在通过临床试验后,FDA对其进行审查,以供批准和商业化[451,452]。此外,临床试验的失败率增加了药物开发过程的低效性,每次失败的试验都会破坏投资并影响临床前试验的成本。高失败率背后的两个主要原因是患者选择不当和试验期间监测效率低下。此外,人工智能技术的引入,大大提高了临床试验的成功率[453]。IBM Watson开发了一个临床试验匹配系统,该系统使用患者的病历和大量过去的临床试验数据来创建详细的临床发现概要。它还可用于检查登记的患者[454]。AI模型还可以通过分析毒性、副作用和其他相关参数来提高成功率,从而降低临床试验的成本[455]。其中一个这样的例子预测了I期和II期临床试验的结果,它基于DL并计算了可能的副作用概率和通路激活评分,进一步用于训练模型[456]。同样,另一个名为“视觉生理人”的项目也被用于支持电子(in silico)试验[457]。此外,人工智能技术的发展将有助于更好地管理临床试验数据,最终开发个性化药物。
10. 人工智能在药物开发中的应用——以神经退行性疾病为例
NDD是致命的、多方面的、使人衰弱的中枢神经系统疾病,是全世界范围死亡的主要原因。AD、PD、肌萎缩侧索硬化症(ALS)和亨廷顿病(HD)是最常见的NDD,最终可导致中枢神经系统不同区域的神经元死亡[458]。毒性、错误折叠的细胞质蛋白在不同脑区的聚集是这些疾病发生的主要原因之一[459]。此外,这些障碍可表现出不同的症状,如认知能力下降、动作迟缓、震颤、记忆力丧失、抑郁、说话问题、肌肉僵硬等[460,461]。NDD带来的主要挑战是在药物发现领域,迄今为止,尚未发现能够阻止和逆转这种疾病进展的药物。因此,迫切需要新的药物靶点和药物化合物,它们可以缓解中枢神经系统的症状和疾病状况[462]。目前,ML被广泛用于寻找与NDD相关的新靶点和生物标志物。例如,Martínez-Ballesteros等人2016将DT、定量关联规则和层次聚类结合起来,通过患者和对照样本的基因表达谱确定AD的潜在风险基因。此外,文献[463]结合蛋白质-蛋白质相互作用网络、自动编码器和支持向量机预测与PD相关的新靶基因,同样地,文献[464]使用诸如RF、DT、广义线性模型和规则归纳等ML模型通过基因表达谱来发现HD的风险基因。此外,文献[465]使用CNN对广泛的GWAS数据集进行训练,以发现新的风险单核苷酸多态性和与ALS相关的基因。
此外,ML技术也被用于寻找NDD相关靶蛋白的合适抑制剂。例如,文献[466]应用VS、ML和分子对接的组合来发现1类和IIb类组蛋白去乙酰化酶抑制剂,因为HDAC酶已被报道促进AD神经毒性。在这里,ML用于抑制剂和非抑制剂后-VS的分类。此外,文献[467]使用来自caspase-8蛋白-配体复合物的MD模拟轨迹的描述符来训练ANN和RF模型,以找到caspase 8蛋白酶的抑制剂,这是一种与AD发病机制有关的蛋白酶。在另一项研究中,文献[468]使用来自中医药数据库的数据,然后使用VS、分子对接和ML技术(包括DL)来寻找GSK3β的抑制剂,GSK3β是一种与AD相关的酶。此外,使用MD模拟来评估GSK3β-配体相互作用的稳定性。此外,Ponzoni等人2019建立了一个QSAR模型,用于寻找BACE1酶的抑制剂,BACE1酶负责AD中β-淀粉样蛋白(aβ)聚集。在这里,QSAR模型是使用一组最佳的分子描述符构建的,这些分子描述符使用ML算法、杂交技术、反向消除策略和视觉分析的融合进行分类[469]。类似地,文献[470]使用一系列朴素贝叶斯网络寻找有效且安全的阿贝尔森(abelson)酪氨酸蛋白激酶1(c-Abl)抑制剂,促进PD的神经保护。同样,Shao等人2018使用SVM算法和基于Tanimoto相似性的聚类集成,然后进行体外实验,以找到A2A腺苷受体和多巴胺D2受体的新拮抗剂,正如已经观察到的,阻断这两个受体会导致PD的神经保护作用[471]。此外,文献[472]实施了分子对接、AI-QSAR和MD模拟,以发现NLR家族pyrin结构域包含3(NLRP3)的抑制剂,这是一种参与PD发病机制的炎性体。在这里,VS和对接用于从中药数据库中筛选化合物,而AI和QSAR模型用于确定化合物的生物活性,然后通过MD模拟评估其结合稳定性[472]。类似地,文献[473]使用分子对接、AI和MD模拟来发现半乳糖凝集素-3抑制剂,半乳糖凝集素-3是HD中与神经炎症相关的蛋白质。在这里,分子对接用于初步筛选,然后通过ML评估化合物的生物活性,并通过MD模拟评估其结合稳定性。此外,不同的研究使用ML算法在NDD中重新调整药物用途。类似地,X. Zeng等人2019开发了一种基于DL的药物再利用工具,称为deepDR(GitHub – ChengF-Lab/deepDR),用于寻找新的AD和PD再利用药物[291]。此外,文献[474]通过使用遗传网络驱动的分类模型,提出替米沙坦作为潜在的AD再利用药物。此外,文献[475]通过整合知识表示学习和ML算法扫描科学文献,提出了PD的药物再利用策略。
11. 未来的挑战和可能的解决办法
目前,制药行业在开发新药时面临的主要挑战是成本增加和效率降低。然而,ML方法和DL的最新发展带来了在药物发现和开发过程中降低成本、提高效率和节省时间的巨大机会。人工智能算法的发展,特别是DL方法的发展,以及体系结构硬件的改进和大数据的易访问性,都预示着人工智能的第三次浪潮。药物开发中的人工智能方法引起了研究者的极大兴趣,许多制药公司与人工智能公司进行了合作。此外,该领域的初创企业数量也在不断增加,到2020年6月达到230家[476]。此外,DL方法通过非线性模型在多个层次上集成数据,这是AI和ML方法的缺点。然而,在多个层次上集成数据使得DL算法具有优势,因为它提供了非常高的准确性和精确度。此外,与AI和ML算法相比,DL提供了更灵活的体系结构,可以为特定问题创建神经网络[477-480]。像自然语言处理、图像和语音识别这样的人工智能应用现在很容易实现,在性能方面已经超过了人类[481]。因此,人工智能可以很好地应用于药物发现过程也就不足为奇了。如今,人工智能被用于药物发现,用于靶点识别、苗头发现、先导优化、ADMET预测和构建临床试验。尽管取得了巨大成功,但仍然存在许多挑战,如高质量数据采集,其中有两个重要问题。首先,由于药物在生物系统中的作用是复杂的,标记不能是二元的;第二,药物发现中可用的数据量与可用的大量信息相比是微乎其微的。因此,需要一个不仅提供数量而且提供高质量数据的团体。在制药行业,开放式数据共享并不常见,Pistoia联盟已主动发起一项运动,鼓励许多公司与其他公司共享数据。他们还打算建立一种统一的数据格式,这在技术上具有挑战性[161]。解决这个问题的一个可能的方法是开发一种能够处理稀疏数据的算法;斯坦福大学已经开发了一种叫做“一次性学习”的方法,该方法基于异构数据预测药物的性质[482]。此外,实验数据的准确性和不确定性可用于建模,即不必建立新的ML技术,可以通过调整大量的超参数来训练现有的超参数,并对其进行优化以获得良好的结果,尽管一些研究表明,可以使用一些合理的参数来启动优化[435]。分子表征也是一个挑战,因为它是模型建立的控制因素之一。最近开发的模型很少从原始数据中学习与任务相关的特征,并将分子表示细化到一个标准。早些时候,药物的重新用途过去只依赖于临床观察。然而,目前包含科学文献、专利和临床试验结果的大量数据可共同用于改进筛选过程。此外,基于DL的VS可以充分利用数据,并减少由于正负数据不平衡而获得的假阳性率。为了开发具有良好ADMET性质和靶向活性的高效药物,先导的优化也是一个挑战;但是,这些参数是独立的,有时相互不兼容。通过分别优化各参数,进一步改进模型,可以解决这一问题。制药公司在招募足够数量的患者进行临床试验方面面临困难。人工智能方法将有助于确定和招募目标患者,并有助于管理收集的数据。对于神经退行性疾病的药物发现,主要的问题是其未知的病理生理,这使得药物鉴定更具挑战性。ML模型的“黑箱”性质是一个额外的挑战,即使专家也无法解释模型是如何得出结果的,也无法理解其背后的生物学机制。此外,不断增加的ML模型数量以及它们声称是最新的,这使得非专业人员束手无策,因为他们无法决定选择哪个模型来解决他们的问题。因此,如果用户和开发人员对标准的客观评估达成一致,然后检查模型的性能,这将是更好的。此外,值得注意的是,大多数国家都没有为那些完全由人工智能技术创造的发明授予专利。此外,使用人工智能技术进行药物发现的公司必须通过强有力的程序为其作品版权,以确保专利权。安全性也是一个主要问题,因为人工智能驱动的个性化医疗需要个人的遗传代码,而个人信息是必需的。最后,处理大数据需要更快的计算速度,据说未来,量子计算机或其他技术将取代目前的超级计算机,这些技术只需几分钟,而不是几小时。虽然人工智能为不同的疾病提供了许多新的靶点和新的化合物,但是通过人工智能产生的化合物进入市场供公众使用的成功案例还没有出现。近年来,基于人工智能的工具首次提出了一种新的靶点及其抑制剂。Insilico medicine,一家生物技术公司,提出了一种新的靶点参与特发性肺纤维化,并通过他们基于AI的工具从头开始制备其新型抑制剂。所鉴定的小分子抑制剂在人细胞和动物模型中显示出良好的效果。2020年12月,Insilico公司提名其小分子抑制剂用于研究新药(IND),并计划在2022年初进行临床试验。如果试验成功,那么将是有史以来第一次通过基于AI的工具提出新的靶点及其抑制剂并获得批准。尽管在药物发现周期中引入人工智能工具存在着一些不可避免的障碍,需要做大量的工作,但毫无疑问,在不久的将来,人工智能将给药物发现和开发过程带来革命性的变化。
我们要感谢德里科技大学的高层管理人员一直以来的支持和指导。
作者的贡献
所有作者已阅读论文并同意提交。PK构思了这个想法。RG、DS、MS、ST准备数据。RG、DS、MS和ST对这项工作的贡献相同。RKA和PK给出了他们的批评性意见并构建了本文。图片艺术作品由RG、RAK和PK完成。论文由PK撰写。
声明
利益冲突:作者声明不存在利益冲突。
参考文献
- Lipinski CF, Maltarollo VG, Oliveira PR et al (2019) Advances and perspectives in applying deep learning for drug design and discovery. Front Robot AI. https://doi.org/10.3389/frobt.2019. 00108
- Hamet P, Tremblay J (2017) Artificial intelligence in medicine.Metabolism. https://doi.org/10.1016/j.metabol.2017.01.011
- Hassanzadeh P, Atyabi F, Dinarvand R (2019) The significance of artificial intelligence in drug delivery system design. Adv Drug Deliv Rev. https://doi.org/10.1016/j.addr.2019.05.001
- Duch W, Swaminathan K, Meller J (2007) Artificial intelligence approaches for rational drug design and discovery. Curr Pharm Des. https://doi.org/10.2174/138161207780765954
- Zhang L, Tan J, Han D, Zhu H (2017) From machine learning to deep learning: progress in machine intelligence for rational drug discovery. Drug Discov Today. https://doi.org/10.1016/j. drudis.2017.08.010
- Jordan AM (2018) Artificial intelligence in drug design–the storm before the calm? ACS Med Chem Lett. https://doi.org/10.1021/acsmedchemlett.8b00500
- Goel AK, Davies J (2019) Artificial intelligence. In: The Cambridge Handbook of Intelligence. Cambridge
- Harrer S, Shah P, Antony B, Hu J (2019) Artificial Intelligence for Clinical Trial Design. Sci, Trends Pharmacol. https://doi.org/10.1016/j.tips.2019.05.005
- Zhong F, Xing J, Li X et al (2018) Artificial intelligence in drug design. Sci China Life Sci. https://doi.org/10.1007/s11427-018-9342-2
- Brown N, Ertl P, Lewis R et al (2020) Artificial intelligence in chemistry and drug design. J Comput Aided Mol Des. https://doi.org/10.1007/s10822-020-00317-x
- Badillo S, Banfai B, Birzele F et al (2020) An introduction to machine learning. Clin Pharmacol Ther. https://doi.org/10.1002/cpt.1796
- Dutta Majumdar D (1985) Trends in pattern recognition and machine learning. Def Sci J. https://doi.org/10.14429/dsj.35.6027
- Kubat M (2017) An Introduction to Machine Learning
- Aggarwal M, Murty MN (2021) Deep Learning. In: SpringerBriefs in Applied Sciences and Technology. https://doi.org/10.1007/978-981-33-4022-0_3
- Schmidhuber J (2015) Deep learning in neural networks: an overview. Neural Netw. https://doi.org/10.1016/j.neunet.2014.09.003
- Hu YH, Hwang JN (2001) Introduction to neural networks for signal processing. In: Handbook of Neural Network Signal Processing. CRC Press, pp 12–41
- Angermueller C, Pärnamaa T, Parts L, Stegle O (2016) Deep learning for computational biology. Mol Syst Biol. https://doi.org/10.15252/msb.20156651
- McCulloch WS, Pitts W (1943) A logical calculus of the ideas immanent in nervous activity. Bull Math Biophys 5:115–133. https://doi.org/10.1007/BF02478259
- Turing AM (2009) Computing machinery and intelligence. Parsing the Turing Test: Philosophical and Methodological Issues in the Quest for the Thinking Computer. Springer, Netherlands, pp 23–65
- Samuel AL (1959) Some studies in machine learning using the game of checkers. IBM J Res Dev 3:210–229. https://doi.org/10.1147/rd.33.0210
- Rosenblatt F (1957) The Perceptron: A Perceiving and Recognizing Automaton, Report 85–60–1
- KELLEY HJ, (1960) Gradient theory of optimal flight paths. ARS J 30:947–954. https://doi.org/10.2514/8.5282
- Dreyfus S (1962) The numerical solution of variational problems. J Math Anal Appl 5:30–45. https://doi.org/10.1016/0022-247X(62)90004-5
- Fukushima K (1980) Neocognitron: a self-organizing neural network model for a mechanism of pattern recognition unaffected by shift in position. Biol Cybern 36:193–202. https://doi.org/10.1007/BF00344251
- Fukushima K (1988) Neocognitron: a hierarchical neural network capable of visual pattern recognition. Neural Netw 1(2):119–130. https://doi.org/10.1016/0893-6080(88)90014-7
- Rumelhart DE, Hinton GE, Williams RJ (1986) Learning representations by back-propagating errors. Nature 323:533–536. https://doi.org/10.1038/323533a0
- LeCun Y, Boser B, Denker JS et al (1989) Backpropagation applied to handwritten zip code recognition. Neural Comput 1:541–551. https://doi.org/10.1162/neco.1989.1.4.541
- Watkins CJCH, Dayan P (1992) Q-learning. Mach Learn 8:279–292. https://doi.org/10.1007/bf00992698
- Cortes C, Vapnik V (1995) Support-vector networks. Mach Learn 20:273–297. https://doi.org/10.1023/A:1022627411411
- Hochreiter S, Schmidhuber J (1997) Long short-term memory. Neural Comput 9:1735–1780. https://doi.org/10.1162/neco. 1997.9.8.1735
- Ilievski A, Zdraveski V, Gusev M (2018) How CUDA Powers the machine learning revolution. 2018 26th Telecommun Forum, TELFOR 2018 – Proc 420–425. https://doi.org/https://doi.org/10.1109/TELFOR.2018.8611982
- Deng J, Dong W, Socher R et al (2010) ImageNet: a large-scale hierarchical image database. Inst Electric Electron Eng IEEE. https://doi.org/10.1109/CVPR.2009.5206848
- Krizhevsky A, Sutskever I, Hinton GE (2012) ImageNet Classification with Deep Convolutional Neural Networks. In Proceedings of the 25th International Conference on Neural Information Processing Systems – Volume 1
- Le Q V, Ranzato M’ A, Monga R, et al (2012) Building Highlevel Features Using Large Scale Unsupervised Learning. https://arxiv.org/abs/1112.6209v5
- Jorda M, Valero-Lara P, Pena AJ (2019) Performance evaluation of cuDNN convolution algorithms on NVIDIA volta GPUs. IEEE Access 7:70461–70473. https://doi.org/10.1109/ACCESS.2019.2918851
- Taigman Y, Yang M, Ranzato M, Wolf L (2014) DeepFace: Closing the gap to human-level performance in face verification. In: Proceedings of the IEEE Computer Society Conference on Computer Vision and Pattern Recognition. IEEE Computer Society, pp 1701–1708
- Goodfellow I, Pouget-Abadie J, Mirza M et al (2020) Generative Adversarial Networks. Commun ACM. https://doi.org/10.1145/3422622
- Gandomi A, Haider M (2015) Beyond the hype: Big data concepts, methods, and analytics. Int J Inf Manage 35:137–144. https://doi.org/10.1016/j.ijinfomgt.2014.10.007
- Brazma A, Kapushesky M, Parkinson H et al (2006) [20] Data Storage and Analysis in ArrayExpress. Methods Enzymol 411:370–86. https://doi.org/10.1016/S0076-6879(06)11020-4
- Lo Y-C, Ren G, Honda H, L. Davis K (2020) Artificial Intelligence-Based Drug Design and Discovery. In: Cheminformatics and its Applications: https://doi.org/10.5772/intechopen.89012
- Edgar R, Domrachev M, Lash AE (2002) Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. https://doi.org/10.1093/nar/30.1.207
- Wang Z, Jensen MA, Zenklusen JC (2016) A practical guide to The Cancer Genome Atlas (TCGA). In: Methods in Molecular Biology 1418:111–41: https://doi.org/10.1007/978-1-4939-3578-9_6
- Parkinson H, Kapushesky M, Shojatalab M et al (2007) ArrayExpress-a public database of microarray experiments and gene expression profiles. Nucleic Acids Res. https://doi.org/10.1093/nar/gkl995
- van IJzendoorn DGP, Szuhai K, Briaire-De Bruijn IH, et al (2019) Machine learning analysis of gene expression data reveals novel diagnostic and prognostic biomarkers and identifies therapeutic targets for soft tissue sarcomas. PLoS Comput Biol 15:1–19. https://doi.org/10.1371/journal.pcbi.1006826
- Lau A, So HC (2020) Turning genome-wide association study findings into opportunities for drug repositioning. Comput Struct Biotechnol J 18:1639–1650. https://doi.org/10.1016/j.csbj.2020.06.015
- Beck T, Hastings RK, Gollapudi S et al (2014) GWAS Central: a comprehensive resource for the comparison and interrogation of genome-wide association studies. Eur J Hum Genet. https://doi.org/10.1038/ejhg.2013.274
- Buniello A, Macarthur JAL, Cerezo M et al (2019) The NHGRIEBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. https://doi.org/10.1093/nar/gky1120
- Li J, Yuan X, March ME et al (2019) Identification of target genes at juvenile idiopathic arthritis GWAS loci in human neutrophils. Front Genet. https://doi.org/10.3389/fgene.2019.00181
- Leinonen R, Sugawara H, Shumway M (2011) The sequence read archive. Nucleic Acids Res. https://doi.org/10.1093/nar/gkq1019
- Jensen MA, Ferretti V, Grossman RL, Staudt LM (2017) The NCI genomic data commons as an engine for precision medicine. Blood 130(4):453–459. https://doi.org/10.1182/blood-2017-03-735654
- Han Y, Yang J, Qian X et al (2019) DriverML: a machine learning algorithm for identifying driver genes in cancer sequencing studies. Nucleic Acids Res. https://doi.org/10.1093/nar/gkz096
- Guillaume JC (1998) PubMed. Ann Dermatol Venereol. https://doi.org/10.1002/9783527678679.dg10319
- Canese K, Weis S (2013) PubMed: The bibliographic database. NCBI Handb
- Kim S, Chen J, Cheng T et al (2021) PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. https://doi.org/10.1093/nar/gkaa971
- Kim S, Chen J, Cheng T et al (2019) PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. https://doi.org/10.1093/nar/gky1033
- Mendez D, Gaulton A, Bento AP et al (2019) ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. https://doi.org/10.1093/nar/gky1075
- Bento AP, Gaulton A, Hersey A et al (2014) The ChEMBL bioactivity database: an update. Nucleic Acids Res. https://doi.org/10.1093/nar/gkt1031
- Wishart DS, Knox C, Guo AC et al (2008) DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. https://doi.org/10.1093/nar/gkm958
- Wishart DS, Feunang YD, Guo AC et al (2018) DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. https://doi.org/10.1093/nar/gkx1037
- Keenan AB, Jenkins SL, Jagodnik KM et al (2018) The library of integrated network-based cellular signatures NIH program: system-level cataloging of human cells response to perturbations. Cell Syst 6(1):13–24. https://doi.org/10.1016/j.cels.2017.11.001
- Duan Q, Reid SP, Clark NR et al (2016) L1000CDS2: LINCS L1000 characteristic direction signatures search engine. npj Syst Biol Appl 2:1–12. https://doi.org/10.1038/npjsba.2016.15
- Rose PW, Prlić A, Altunkaya A et al (2017) The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw1000
- Burley SK, Berman HM, Bhikadiya C et al (2019) RCSB Protein data bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. https://doi.org/10.1093/nar/gky1004
- Xu Z, Yang L, Zhang X et al (2020) Discovery of potential flavonoid inhibitors against COVID-19 3CL proteinase based on virtual screening strategy. Front Mol Biosci 7:1–8. https://doi.org/10.3389/fmolb.2020.556481
- Fan Y, Zhang Y, Hua Y et al (2019) Investigation of machine intelligence in compound cell activity classification. Mol Pharm. https://doi.org/10.1021/acs.molpharmaceut.9b00558
- Chi CT, Lee MH, Weng CF, Leong MK (2019) In silico prediction of PAMPA effective permeability using a two-QSAR approach. Int J Mol Sci. https://doi.org/10.3390/ijms20133170
- He S, Zhang X, Lu S et al (2019) A computational toxicology approach to screen the hepatotoxic ingredients in traditional chinese medicines: polygonum multiflorum thunb as a case study. Biomolecules. https://doi.org/10.3390/biom9100577
- He S, Zhang C, Zhou P et al (2019) Herb-induced liver injury: Phylogenetic relationship, structure-toxicity relationship, and herb-ingredient network analysis. Int. J Mol Sci. 20(15):3633. https://doi.org/10.3390/ijms20153633
- Zhang D, hai, Wu K lun, Zhang X, et al (2020) In silico screening of Chinese herbal medicines with the potential to directly inhibit 2019 novel coronavirus. J Integr Med. https://doi.org/10.1016/j.joim.2020.02.005
- Baldi A (2010) Computational approaches for drug design and discovery: an overview. Syst Rev Pharm 1(1):99. https://doi.org/10.4103/0975-8453.59519
- Lavecchia A, Cerchia C (2016) In silico methods to address polypharmacology: current status, applications and future perspectives. Drug Discov Today 21(2):288–298. https://doi.org/10.1016/j.drudis.2015.12.007
- Smith JS, Roitberg AE, Isayev O (2018) Transforming computational drug discovery with machine learning and AI. ACS Med Chem Lett 9(11):1065–1069. https://doi.org/10.1021/acsmedchemlett.8b00437
- Jing Y, Bian Y, Hu Z et al (2018) Deep learning for drug design: an artificial intelligence paradigm for drug discovery in the big data era. AAPS J 20(3):58. https://doi.org/10.1208/s12248-018-0210-0
- Powles J, Hodson H (2017) Google deepmind and healthcare in an age of algorithms. Health Technol (Berl). https://doi.org/10.1007/s12553-017-0179-1
- Senior AW, Evans R, Jumper J et al (2020) Improved protein structure prediction using potentials from deep learning. Nature 577:706–710. https://doi.org/10.1038/s41586-019-1923-7
- AlQuraishi M (2019) End-to-End differentiable learning of protein structure. Cell Syst 8:292-301.e3. https://doi.org/10.1016/j.cels.2019.03.006
- Kalaiarasi C, Manjula S, Kumaradhas P (2019) Combined quantum mechanics/molecular mechanics (QM/MM) methods to understand the charge density distribution of estrogens in the active site of estrogen receptors. RSC Adv. https://doi.org/10.1039/c9ra08607b
- Schütt KT, Gastegger M, Tkatchenko A et al (2019) Unifying machine learning and quantum chemistry with a deep neural network for molecular wavefunctions. Nat Commun. https://doi.org/10.1038/s41467-019-12875-2
- Gastegger M, McSloy A, Luya M et al (2020) A deep neural network for molecular wave functions in quasi-atomic minimal basis representation. J Chem Phys DOI. https://doi.org/10.1063/5.0012911
- De Vivo M, Masetti M, Bottegoni G, Cavalli A (2016) Role of molecular dynamics and related methods in drug discovery. J Med Chem 59(9):4035–4061. https://doi.org/10.1021/acs.jmedchem.5b01684
- Bennett WFD, He S, Bilodeau CL et al (2020) Predicting small molecule transfer free energies by combining molecular dynamics simulations and deep learning. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.0c00318
- Bai Q, Tan S, Xu T et al (2020) MolAICal: a soft tool for 3D drug design of protein targets by artificial intelligence and classical algorithm. Brief Bioinform 00:1–12. https://doi.org/10.1093/bib/bbaa161
- Sterling T, Irwin JJ (2015) ZINC 15-ligand discovery for everyone. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.5b00559
- Popova M, Isayev O, Tropsha A (2018) Deep reinforcement learning for de novo drug design. Sci Adv 4:1–15. https://doi.org/10.1126/sciadv.aap7885
- Grzybowski BA, Szymkuć S, Gajewska EP et al (2018) Chematica: a story of computer code that started to think like a chemist. Chem 4:390–398. https://doi.org/10.1016/j.chempr.2018.02.024
- Genheden S, Thakkar A, Chadimová V et al (2020) AiZynthFinder: a fast, robust and flexible open-source software for retrosynthetic planning. J Cheminform 12:1–9. https://doi.org/10.1186/s13321-020-00472-1
- Segler MHS, Preuss M, Waller MP (2018) Planning chemical syntheses with deep neural networks and symbolic AI. Nature 555:604–610. https://doi.org/10.1038/nature25978
- Bøgevig A, Federsel HJ, Huerta F et al (2015) Route design in the 21st century: the IC SYNTH software tool as an idea generator for synthesis prediction. Org Process Res Dev 19:357–368. https://doi.org/10.1021/op500373e
- Jang G, Lee T, Hwang S et al (2018) PISTON: predicting drug indications and side effects using topic modeling and natural language processing. J Biomed Inform 87:96–107. https://doi.org/10.1016/j.jbi.2018.09.015
- Piñero J, Bravo Á, Queralt-Rosinach N et al (2017) DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw943
- Szklarczyk D, Gable AL, Lyon D et al (2019) STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. https://doi.org/10.1093/nar/gky1131
- Szklarczyk D, Santos A, Von Mering C et al (2016) STITCH 5: augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res 44:D380–D384. https://doi.org/10.1093/nar/gkv1277
- Davenport TH, Ronanki R (2018) Artificial intelligence for the real world. Harv Bus Rev
- Zhavoronkov A, Vanhaelen Q, Oprea TI (2020) Will Artificial Intelligence for Drug Discovery Impact Clinical Pharmacology? Clin Pharmacol Ther. https://doi.org/10.1002/cpt.1795
- Watson O, Cortes-Ciriano I, Taylor A, Watson JA (2018) A decision theoretic approach to model evaluation in computational drug discovery. arXiv. https://arxiv.org/abs/1807.08926
- Tripathy RK, Mahanta S, Paul S (2014) Artificial intelligencebased classification of breast cancer using cellular images. RSC Adv 4:9349–9355. https://doi.org/10.1039/c3ra47489e
- Samui P, Kothari DP (2011) Utilization of a least square support vector machine (LSSVM) for slope stability analysis. Sci Iran 18:53–58. https://doi.org/10.1016/j.scient.2011.03.007
- Chan HCS, Shan H, Dahoun T et al (2019) Advancing Drug Discovery via Artificial Intelligence. Trends Pharmacol Sci 40:592–604. https://doi.org/10.1016/j.tips.2019.06.004
- Ho CWL, Soon D, Caals K, Kapur J (2019) Governance of automated image analysis and artificial intelligence analytics in healthcare. Clin Radiol 74:329–337. https://doi.org/10.1016/j.crad.2019.02.005
- Andrysek T (2003) Impact of physical properties of formulations on bioavailability of active substance: Current and novel drugs with cyclosporine. In: Molecular Immunology; 39(17–18):1061–5. https://doi.org/10.1016/s0161-5890(03)00077-4.
- Elton DC, Boukouvalas Z, Butrico MS et al (2018) Applying machine learning techniques to predict the properties of energetic materials. Sci Rep 8:9059. https://doi.org/10.1038/s41598-018-27344-x
- Tyrchan C, Evertsson E (2017) Matched molecular pair analysis in short: algorithms, applications and limitations. Comput Struct Biotechnol J 15:86–90. https://doi.org/10.1016/j.csbj.2016.12.003
- Turk S, Merget B, Rippmann F, Fulle S (2017) Coupling matched molecular pairs with machine learning for virtual compound optimization. J Chem Inf Model 57:3079–3085. https://doi.org/10.1021/acs.jcim.7b00298
- Carpenter KA, Huang X (2018) Machine learning-based virtual screening and its applications to Alzheimer’s drug discovery: a review. Curr Pharm Des 24:3347–3358. https://doi.org/10.2174/1381612824666180607124038
- Schyman P, Liu R, Desai V, Wallqvist A (2017) vNN web server for ADMET predictions. Front Pharmacol 8:889. https://doi.org/10.3389/fphar.2017.00889
- Álvarez-Machancoses Ó, Fernández-Martínez JL (2019) Using artificial intelligence methods to speed up drug discovery. Expert Opin Drug Discov 14(8):769–777. https://doi.org/10.1080/17460441.2019.1621284
- Fleming N (2018) How artificial intelligence is changing drug discovery. Nature. How artificial intelligence is changing drug discovery
- Segler MHS, Kogej T, Tyrchan C, Waller MP (2018) Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent Sci. https://doi.org/10.1021/acscentsci.7b00512
- Bruno BJ, Miller GD, Lim CS (2013) Basics and recent advances in peptide and protein drug delivery. Ther. Deliv 4(11):1443–67. https://doi.org/10.4155/tde.13.104
- Yan J, Bhadra P, Li A et al (2020) Deep-AmPEP30: improve short antimicrobial peptides prediction with deep learning. Mol Ther-Nucleic Acids 20:882–894. https://doi.org/10.1016/j.omtn. 2020.05.006
- Plisson F, Ramírez-Sánchez O, Martínez-Hernández C (2020) Machine learning-guided discovery and design of non-hemolytic peptides. Sci Rep 10:1–19. https://doi.org/10.1038/s41598-020-73644-6
- Kavousi K, Bagheri M, Behrouzi S et al (2020) IAMPE: NMRassisted computational prediction of antimicrobial peptides. J Chem Inf Model 60:4691–4701. https://doi.org/10.1021/acs.jcim.0c00841
- Yi HC, You ZH, Zhou X et al (2019) ACP-DL: a deep learning long short-term memory model to predict anticancer peptides using high-efficiency feature representation. Mol Ther-Nucleic Acids 17:1–9. https://doi.org/10.1016/j.omtn.2019.04.025
- Yu L, Jing R, Liu F et al (2020) DeepACP: a novel computational approach for accurate identification of anticancer peptides by deep learning algorithm. Mol Ther-Nucleic Acids 22:862–870. https://doi.org/10.1016/j.omtn.2020.10.005
- Tyagi A, Kapoor P, Kumar R et al (2013) In silico models for designing and discovering novel anticancer peptides. Sci Rep 3:1–8. https://doi.org/10.1038/srep02984
- Rao B, Zhang L, Zhang G (2020) ACP-GCN: the identification of anticancer peptides based on graph convolution networks. IEEE Access 8:176005–176011. https://doi.org/10.1109/access.2020.3023800
- Wu C, Gao R, Zhang Y, De Marinis Y (2019) PTPD: predicting therapeutic peptides by deep learning and word2vec. BMC Bioinformatics 20:1–8. https://doi.org/10.1186/s12859-019-3006-z
- Zhavoronkov A, Ivanenkov YA, Aliper A et al (2019) Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat Biotechnol 37:1038–1040. https://doi.org/10.1038/s41587-019-0224-x
- McCloskey K, Sigel EA, Kearnes S et al (2020) Machine learning on DNA-encoded libraries: a new paradigm for hit finding. J Med Chem 63:8857–8866. https://doi.org/10.1021/acs.jmedchem.0c00452
- Xing G, Liang L, Deng C et al (2020) Activity prediction of small molecule inhibitors for antirheumatoid arthritis targets based on artificial intelligence. ACS Comb Sci. https://doi.org/10.1021/acscombsci.0c00169
- Dimmitt S, Stampfer H, Martin JH (2017) When less is more–efficacy with less toxicity at the ED50. Br J Clin Pharmacol 83(7):1365–1368. https://doi.org/10.1111/bcp.13281
- Shen Y, Liu T, Chen J et al (2020) Harnessing artificial intelligence to optimize long-term maintenance dosing for antiretroviral-naive adults with HIV-1 Infection. Adv Ther 3:1900114. https://doi.org/10.1002/adtp.201900114
- Pantuck AJ, Lee D-K, Kee T et al (2018) Modulating BET bromodomain inhibitor ZEN-3694 and Enzalutamide combination dosing in a metastatic prostate cancer patient using CURATE. AI an artificial intelligence platform. Adv Ther. https://doi.org/ 10.1002/adtp.201800104
- Julkunen H, Cichonska A, Gautam P et al (2020) Leveraging multi-way interactions for systematic prediction of pre-clinical drug combination effects. Nat Commun. https://doi.org/10.1038/s41467-020-19950-z
- Sharabiani A, Bress A, Douzali E, Darabi H (2015) Revisiting warfarin dosing using machine learning techniques. Comput Math Methods Med. https://doi.org/10.1155/2015/560108
- Nemati S, Ghassemi MM, Clifford GD (2016) Optimal medication dosing from suboptimal clinical examples: a deep reinforcement learning approach. Proc Annu Int Conf IEEE Eng Med Biol Soc EMBS. https://doi.org/10.1109/EMBC.2016.7591355
- Tang J, Liu R, Zhang YL et al (2017) Application of machinelearning models to predict tacrolimus stable dose in renal transplant recipients. Sci Rep. https://doi.org/10.1038/srep42192
- Hu YH, Tai CT, Tsai CF, Huang MW (2018) Improvement of adequate digoxin dosage: an application of machine learning approach. J Healthc Eng. https://doi.org/10.1155/2018/3948245
- Imai S, Takekuma Y, Miyai T, Sugawara M (2020) A new algorithm optimized for initial dose settings of vancomycin using machine learning. Biol Pharm Bull 43:188–193. https://doi.org/10.1248/bpb.b19-00729
- Rollinger JM, Stuppner H, Langer T (2008) Virtual screening for the discovery of bioactive natural products. Prog Drug Res 65:212–249. https://doi.org/10.1007/978-3-7643-8117-2_6
- Schuster D, Maurer EM, Laggner C et al (2006) The discovery of new 11β-hydroxysteroid dehydrogenase type 1 inhibitors by common feature pharmacophore modeling and virtual screening. J Med Chem 49:3454–3466. https://doi.org/10.1021/jm0600794
- Wu J, Zhang Q, Wu W et al (2018) WDL-RF: predicting bioactivities of ligand molecules acting with G protein-coupled receptors by combining weighted deep learning and random forest. Bioinformatics 34:2271–2282. https://doi.org/10.1093/bioinformatics/bty070
- Cichonska A, Pahikkala T, Szedmak S et al (2018) Learning with multiple pairwise kernels for drug bioactivity prediction. Bioinformatics 34:i509–i518. https://doi.org/10.1093/bioinformatics/bty277
- Babajide Mustapha I, Saeed F (2016) Bioactive molecule prediction using extreme gradient boosting. Molecules 21:1–11. https://doi.org/10.3390/molecules21080983
- Merget B, Turk S, Eid S et al (2017) Profiling prediction of kinase inhibitors: toward the virtual assay. J Med Chem 60:474–485. https://doi.org/10.1021/acs.jmedchem.6b01611
- Arshadi AK, Salem M, Collins J et al (2020) Deepmalaria: artificial intelligence driven discovery of potent antiplasmodials. Front Pharmacol. https://doi.org/10.3389/fphar.2019.0152
- Sugaya N (2014) Ligand efficiency-based support vector regression models for predicting bioactivities of ligands to drug target proteins. J Chem Inf Model 54:2751–2763. https://doi.org/10.1021/ci5003262
- Afolabi LT, Saeed F, Hashim H, Petinrin OO (2018) Ensemble learning method for the prediction of new bioactive molecules. PLoS ONE 13:1–14. https://doi.org/10.1371/journal.pone.0189538
- Petinrin OO, Saeed F (2018) Bioactive molecule prediction using majority voting-based ensemble method. J Intell Fuzzy Syst 35:383–392. https://doi.org/10.3233/JIFS-169596
- Liu X, Gao Y, Peng J et al (2015) TarPred: a web application for predicting therapeutic and side effect targets of chemical compounds. Bioinformatics. https://doi.org/10.1093/bioinformatics/btv099
- Liu M, Wu Y, Chen Y et al (2012) Large-scale prediction of adverse drug reactions using chemical, biological, and phenotypic properties of drugs. J Am Med Informatics Assoc 19:28–35. https://doi.org/10.1136/amiajnl-2011-000699
- Jamal S, Goyal S, Shanker A, Grover A (2017) Predicting neurological adverse drug reactions based on biological, chemical and phenotypic properties of drugs using machine learning models. Sci Rep 7:1–12. https://doi.org/10.1038/s41598-017-00908-z
- Xue R, Liao J, Shao X et al (2020) Prediction of adverse drug reactions by combining biomedical tripartite network and graph representation model. Chem Res Toxicol 33:202–210. https://doi.org/10.1021/acs.chemrestox.9b00238
- Raja K, Patrick M, Elder JT, Tsoi LC (2017) Machine learning workflow to enhance predictions of adverse drug reactions (ADRs) through drug-gene interactions: application to drugs for cutaneous diseases. Sci Rep 7:1–11. https://doi.org/10.1038/s41598-017-03914-3
- Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. https://doi.org/10.1038/srep42717
- Rost B, Liu J, Nair R et al (2003) Automatic prediction of protein function. Cell Mol Life Sci 60:2637–2650. https://doi.org/10.1007/s00018-003-3114-8
- Browne F, Zheng H, Wang H, Azuaje F (2010) From experimental approaches to computational techniques: a review on the prediction of protein-protein interactions. Adv Artif Intell. https://doi.org/10.1155/2010/924529
- Hale WH (1913) American association for the advancement of science. Sci Am 75:34–34. https://doi.org/10.1038/scientificamerican01181913-34supp
- Troyanskaya OG, Dolinski K, Owen AB et al (2003) A Bayesian framework for combining heterogeneous data sources for gene function prediction (in Saccharomyces cerevisiae). Proc Natl Acad Sci U S A 100:8348–8353. https://doi.org/10.1073/pnas.0832373100
- You ZH, Lei YK, Zhu L et al (2013) Prediction of proteinprotein interactions from amino acid sequences with ensemble extreme learning machines and principal component analysis. BMC Bioinformatics 14:1–11. https://doi.org/10.1186/1471-2105-14-S8-S10
- Du X, Sun S, Hu C et al (2017) DeepPPI: boosting prediction of protein-protein interactions with deep neural networks. J Chem Inf Model 57:1499–1510. https://doi.org/10.1021/acs.jcim.7b00028
- Cunningham JM, Koytiger G, Sorger PK, AlQuraishi M (2020) Biophysical prediction of protein–peptide interactions and signaling networks using machine learning. Nat Methods 17:175–183. https://doi.org/10.1038/s41592-019-0687-1
- Chatterjee P, Basu S, Kundu M et al (2011) PPI_SVM: prediction of protein-protein interactions using machine learning, domain-domain affinities and frequency tables. Cell Mol Biol Lett 16:264–278. https://doi.org/10.2478/s11658-011-0008-x
- Lu L, Lu H, Skolnick J (2002) Multiprospector: an algorithm for the prediction of protein-protein interactions by multimeric threading. Proteins Struct Funct Genet 49:350–364. https://doi.org/10.1002/prot.10222
- Singh R, Park D, Xu J et al (2010) Struct2Net: a web service to predict protein-protein interactions using a structure-based approach. Nucleic Acids Res 38:508–515. https://doi.org/10.1093/nar/gkq481
- Dandekar T, Snel B, Huynen M, Bork P (1998) Conservation of gene order: a fingerprint of proteins that physically interact. Trends Biochem Sci 23:324–328. https://doi.org/10.1016/S0968-0004(98)01274-2
- Keskin O, Tuncbag N, Gursoy A (2016) Predicting protein-protein interactions from the molecular to the proteome level. Chem Rev 116:4884–4909. https://doi.org/10.1021/acs.chemrev.5b00683
- Lavecchia A, Giovanni C (2013) Virtual screening strategies in drug discovery: a critical review. Curr Med Chem. https://doi.org/10.2174/09298673113209990001
- Gonczarek A, Tomczak JM, Zaręba S et al (2018) Interaction prediction in structure-based virtual screening using deep learning. Comput Biol Med. https://doi.org/10.1016/j.compbiomed. 2017.09.007
- Goh GB, Hodas NO, Vishnu A (2017) Deep learning for computational chemistry. J Comput Chem 38(16):1291–1307. https://doi.org/10.1002/jcc.24764
- Yang X, Wang Y, Byrne R et al (2019) Concepts of artificial intelligence for computer-assisted drug discovery. Chem. Rev 119(18):10520–10594. https://doi.org/10.1021/acs.chemrev.8b00728
- Arciniega M, Lange OF (2014) Improvement of virtual screening results by docking data feature analysis. J Chem Inf Model. https://doi.org/10.1021/ci500028u
- Feinstein WP, Brylinski M (2015) Calculating an optimal box size for ligand docking and virtual screening against experimental and predicted binding pockets. J Cheminform. https://doi.org/10.1186/s13321-015-0067-5
- Gazgalis D, Zaka M, Zaka M et al (2020) Protein binding pocket optimization for virtual high-throughput screening (vHTS) drug discovery. ACS Omega. https://doi.org/10.1021/acsomega.0c00522
- Carpenter KA, Huang X (2018) Machine learning-based virtual screening and its applications to Alzheimer’s drug discovery: a review. Curr Pharm Des. https://doi.org/10.2174/1381612824666180607124038
- Serafim MSM, Kronenberger T, Oliveira PR et al (2020) The application of machine learning techniques to innovative antibacterial discovery and development. Expert Opin Drug Discov. https://doi.org/10.1080/17460441.2020.1776696
- Melville J, Burke E, Hirst J (2009) Machine learning in virtual screening. Comb Chem High Throughput Screen. https://doi.org/10.2174/138620709788167980
- Wójcikowski M, Ballester PJ, Siedlecki P (2017) Performance of machine-learning scoring functions in structure-based virtual screening. Sci Rep. https://doi.org/10.1038/srep46710
- Carpenter KA, Cohen DS, Jarrell JT, Huang X (2018) Deep learning and virtual drug screening. Future Med Chem. 10(21):2557–2567. https://doi.org/10.4155/fmc-2018-0314
- Labbé CM, Rey J, Lagorce D et al (2015) MTiOpenScreen: a web server for structure-based virtual screening. Nucleic Acids Res. https://doi.org/10.1093/nar/gkv306
- Schellhammer I, Rarey M (2004) FlexX-Scan: Fast, structurebased virtual screening. Proteins Struct Funct Bioinforma 57:504–517. https://doi.org/10.1002/prot.20217
- Perez-Castillo Y, Sotomayor-Burneo S, Jimenes-Vargas K et al (2019) CompScore: boosting structure-based virtual screening performance by incorporating docking scoring function components into consensus scoring. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.9b00343
- Skalic M, Martínez-Rosell G, Jiménez J, De Fabritiis G (2019) PlayMolecule bindscope: large scale CNN-based virtual screening on the web. Bioinformatics. https://doi.org/10.1093/bioinformatics/bty758
- Fang Y, Ding Y, Feinstein WP et al (2016) GeauxDock: accelerating structure-based virtual screening with heterogeneous computing. PLoS ONE. https://doi.org/10.1371/journal.pone.0158898
- Pires DEV, Veloso WNP, Myung YC et al (2020) EasyVS: a user-friendly web-based tool for molecule library selection and structure-based virtual screening. Bioinformatics. https://doi.org/10.1093/bioinformatics/btaa480
- Ibrahim TM, Bauer MR, Boeckler FM (2015) Applying DEKOIS 2.0 in structure-based virtual screening to probe the impact of preparation procedures and score normalization. J Cheminform. https://doi.org/10.1186/s13321-015-0074-6
- Shin WH, Christoffer CW, Wang J, Kihara D (2016) PL-PatchSurfer2: improved local surface matching-based virtual screening method that is tolerant to target and ligand structure variation. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.6b00163
- Litfin T, Zhou Y, Yang Y (2017) SPOT-ligand 2: improving structure-based virtual screening by binding-homology search on an expanded structural template library. Bioinformatics. https://doi.org/10.1093/bioinformatics/btw829
- Ropp PJ, Spiegel JO, Walker JL et al (2019) GypSUm-DL: An open-source program for preparing small-molecule libraries for structure-based virtual screening. J Cheminform. https://doi.org/10.1186/s13321-019-0358-3
- Akbar R, Jusoh SA, Amaro RE, Helms V (2017) ENRI: a tool for selecting structure-based virtual screening target conformations. Chem Biol Drug Des. https://doi.org/10.1111/cbdd.12900
- Kellenberger E, Springael JY, Parmentier M et al (2007) Identification of nonpeptide CCR5 receptor agonists by structure-based virtual screening. J Med Chem. https://doi.org/10.1021/jm061389p
- De Graaf C, Rognan D (2008) Selective structure-based virtual screening for full and partial agonists of the β2 adrenergic receptor. J Med Chem. https://doi.org/10.1021/jm800710x
- Vidler LR, Filippakopoulos P, Fedorov O et al (2013) Discovery of novel small-molecule inhibitors of BRD4 using structurebased virtual screening. J Med Chem. https://doi.org/10.1021/jm4011302
- Liu LJ, Leung KH, Chan DSH et al (2014) Identification of a natural product-like STAT3 dimerization inhibitor by structurebased virtual screening. Cell Death Dis. https://doi.org/10.1038/cddis.2014.250
- Yang C, Wang W, Chen L et al (2016) Discovery of a VHL and HIF1α interaction inhibitor with: in vivo angiogenic activity via structure-based virtual screening. Chem Commun. https://doi.org/10.1039/c6cc04938a
- Zhuang C, Narayanapillai S, Zhang W et al (2014) Rapid identification of Keap1-Nrf2 small-molecule inhibitors through structure-based virtual screening and hit-based substructure search. J Med Chem. https://doi.org/10.1021/jm4017174
- Dou X, Jiang L, Wang Y et al (2018) Discovery of new GSK-3β inhibitors through structure-based virtual screening. Bioorganic Med Chem Lett. https://doi.org/10.1016/j.bmcl.2017.11.036
- Liu Y, Ren Y, Cao Y et al (2017) Discovery of a low toxicity O-GlcNAc Transferase (OGT) inhibitor by structure-based virtual screening of natural products. Sci Rep. https://doi.org/10.1038/s41598-017-12522-0
- Wang Y, Dou X, Jiang L et al (2019) Discovery of novel glycogen synthase kinase-3α inhibitors: Structure-based virtual screening, preliminary SAR and biological evaluation for treatment of acute myeloid leukemia. Eur J Med Chem. https://doi.org/10.1016/j.ejmech.2019.03.039
- Wang Q, Xu J, Li Y et al (2018) Identification of a novel protein arginine methyltransferase 5 inhibitor in non-small cell lung cancer by structure-based virtual screening. Front Pharmacol. https://doi.org/10.3389/fphar.2018.00173
- Sharma K, Patidar K, Ali MA et al (2018) Structure-based virtual screening for the identification of high affinity compounds as potent vegfr2 inhibitors for the treatment of renal cell carcinoma. Curr Top Med Chem. https://doi.org/10.2174/1568026619666181130142237
- Yousuf Z, Iman K, Iftikhar N, Mirza MU (2017) Structure-based virtual screening and molecular docking for the identification of potential multi-targeted inhibitors against breast cancer. Breast Cancer Targets Ther. https://doi.org/10.2147/BCTT.S132074
- Leão M, Pereira C, Bisio A et al (2013) Discovery of a new small-molecule inhibitor of p53-MDM2 interaction using a yeast-based approach. Biochem Pharmacol. https://doi.org/10.1016/j.bcp.2013.01.032
- Gahlawat A, Kumar N, Kumar R et al (2020) Structure-based virtual screening to discover potential lead molecules for the SARS-CoV-2 main protease. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.0c00546
- Selvaraj C, Dinesh DC, Panwar U et al (2020) Structure-based virtual screening and molecular dynamics simulation of SARSCoV-2 guanine-N7 methyltransferase (nsp14) for identifying antiviral inhibitors against COVID-19. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1778535
- Cruz JV, Neto MFA, Silva LB et al (2018) Identification of novel protein kinase receptor type 2 inhibitors using pharmacophore and structure-based virtual screening. Molecules. https://doi.org/10.3390/molecules23020453
- Kannan S, Melesina J, Hauser AT et al (2014) Discovery of inhibitors of schistosoma mansoni hdac8 by combining homology modeling, virtual screening, and in vitro validation. J Chem Inf Model. https://doi.org/10.1021/ci5004653
- Zoete V, Daina A, Bovigny C, Michielin O (2016) SwissSimilarity: a web tool for low to ultra high throughput ligand-based virtual screening. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.6b00174
- Imbernón B, Cecilia JM, Pérez-Sánchez H, Giménez D (2018) METADOCK: a parallel metaheuristic schema for virtual screening methods. Int J High Perform Comput Appl. https://doi.org/10.1177/1094342017697471
- Riniker S, Landrum GA (2013) Open-source platform to benchmark fingerprints for ligand-based virtual screening. J Cheminform. https://doi.org/10.1186/1758-2946-5-26
- Li H, Leung KS, Wong MH, Ballester PJ (2016) USR-VS: a web server for large-scale prospective virtual screening using ultrafast shape recognition techniques. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw320
- Suzuki SD, Ohue M, Akiyama Y (2018) PKRank: a novel learning-to-rank method for ligand-based virtual screening using pairwise kernel and RankSVM. Artif Life Robot. https://doi.org/10.1007/s10015-017-0416-8
- Patel H, Brinkjost T, Koch O (2017) PyGOLD: a python based API for docking based virtual screening workflow generation. Bioinformatics. https://doi.org/10.1093/bioinformatics/btx197
- Banegas-Luna AJ, Cerón-Carrasco JP, Puertas-Martín S, PérezSánchez H (2019) BRUSELAS: HPC generic and customizable software architecture for 3D ligand-based virtual screening of large molecular databases. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.9b00279
- Wang L, Pang X, Li Y et al (2017) RADER: a rapid decoy retriever to facilitate decoy based assessment of virtual screening. Bioinformatics. https://doi.org/10.1093/bioinformatics/btw783
- Mochizuki M, Suzuki SD, Yanagisawa K et al (2019) QEX: target-specific druglikeness filter enhances ligand-based virtual screening. Mol Divers. https://doi.org/10.1007/s11030-018-9842-3
- Zhang H, Liao L, Cai Y et al (2019) IVS2vec: a tool of inverse virtual screening based on word2vec and deep learning techniques. Methods. https://doi.org/10.1016/j.ymeth.2019.03.012
- Arcon JP, Modenutti CP, Avendaño D et al (2019) AutoDock Bias: improving binding mode prediction and virtual screening using known protein-ligand interactions. Bioinformatics. https://doi.org/10.1093/bioinformatics/btz152
- Ebejer JP, Finn PW, Wong WK et al (2019) Ligity: a non-superpositional, knowledge-based approach to virtual screening. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.8b00779
- Zhu Z, Wang X, Yang Y et al (2020) D3Similarity: a ligandbased approach for predicting drug targets and for virtual screening of active compounds against COVID-19. ChemRxiv. https://doi.org/10.26434/chemrxiv.11959323.v1
- Bharti DR, Hemrom AJ, Lynn AM (2019) GCAC: Galaxy workflow system for predictive model building for virtual screening. BMC Bioinformatics. https://doi.org/10.1186/s12859-018-2492-8
- Kong Y, Bender A, Yan A (2018) Identification of Novel Aurora Kinase A (AURKA) Inhibitors via Hierarchical Ligand-Based Virtual Screening. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.7b00300
- Musumeci D, Amato J, Zizza P et al (2017) Tandem application of ligand-based virtual screening and G4-OAS assay to identify novel G-quadruplex-targeting chemotypes. Biochim Biophys Acta – Gen Subj. https://doi.org/10.1016/j.bbagen.2017.01.024
- Yu M, Gu Q, Xu J (2018) Discovering new PI3Kα inhibitors with a strategy of combining ligand-based and structure-based virtual screening. J Comput Aided Mol Des. https://doi.org/10.1007/s10822-017-0092-8
- Halim SA, Khan S, Khan A et al (2017) Targeting dengue virus NS-3 Helicase by Ligand based Pharmacophore Modeling and structure based virtual screening. Front Chem. https://doi.org/10.3389/fchem.2017.00088
- Debnath S, Debnath T, Bhaumik S et al (2019) Discovery of novel potential selective HDAC8 inhibitors by combine ligandbased, structure-based virtual screening and in-vitro biological evaluation. Sci Rep. https://doi.org/10.1038/s41598-019-53376-y
- Fu Y, Sun YN, Yi KH et al (2017) 3D pharmacophore-based virtual screening and docking approaches toward the discovery of novel HPPD inhibitors. Molecules. https://doi.org/10.3390/molecules22060959
- Krishna S, Shukla S, Lakra AD et al (2017) Identification of potent inhibitors of DNA methyltransferase 1 (DNMT1) through a pharmacophore-based virtual screening approach. J Mol Graph Model. https://doi.org/10.1016/j.jmgm.2017.05.014
- Pérez-Nueno VI, Pettersson S, Ritchie DW et al (2009) Discovery of novel HIV entry inhibitors for the CXCR4 receptor by prospective virtual screening. J Chem Inf Model. https://doi.org/10.1021/ci800468q
- Hofmarcher M, Mayr A, Rumetshofer E et al (2020) Large-scale ligand-based virtual screening for SARS-CoV-2 inhibitors using deep neural networks. SSRN Electron J. https://doi.org/10.2139/ssrn.3561442
- Amin SA, Ghosh K, Gayen S, Jha T (2020) Chemical-informatics approach to COVID-19 drug discovery: monte carlo based QSAR, virtual screening and molecular docking study of some in-house molecules as papain-like protease (PLpro) inhibitors. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1780946
- Ferraz WR, Gomes RA, Novaes ALS, Goulart Trossini GH (2020) Ligand and structure-based virtual screening applied to the SARS-CoV-2 main protease: an in silico repurposing study. Future Med Chem. https://doi.org/10.4155/fmc-2020-0165
- Choudhary S, Malik YS, Tomar S (2020) Identification of SARSCoV-2 Cell entry inhibitors by drug repurposing using in silico structure-based virtual screening approach. Front Immunol. https://doi.org/10.3389/fimmu.2020.01664
- Xiao T, Qi X, Chen Y, Jiang Y (2018) Development of Ligandbased big data deep neural network models for virtual screening of large compound libraries. Mol Inform. https://doi.org/10.1002/minf.201800031
- Hu J, Liu Z, Yu DJ, Zhang Y (2018) LS-align: An atom-level, flexible ligand structural alignment algorithm for high-throughput virtual screening. In: Bioinformatics 34(13): 2209–2218; https://doi.org/https://doi.org/10.1093/bioinformatics/bty081
- Ha EJ, Lwin CT, Durrant JD (2020) LigGrep: a tool for filtering docked poses to improve virtual-screening hit rates. J Cheminform. https://doi.org/10.1186/s13321-020-00471-2
- Spiegel JO, Durrant JD (2020) AutoGrow4: an open-source genetic algorithm for de novo drug design and lead optimization. J Cheminform. https://doi.org/10.1186/s13321-020-00429-4
- Chen P, Ke Y, Lu Y et al (2019) Dligand2: an improved knowledge-based energy function for protein–ligand interactions using the distance-scaled, finite, ideal-gas reference state. J Cheminform. https://doi.org/10.1186/s13321-019-0373-4
- Gattani S, Mishra A, Hoque MT (2019) StackCBPred: a stacking based prediction of protein-carbohydrate binding sites from sequence. Carbohydr Res. https://doi.org/10.1016/j.carres.2019.107857
- Li X, Yan X, Yang Y et al (2019) LSA: a local-weighted structural alignment tool for pharmaceutical virtual screening. RSC Adv. https://doi.org/10.1039/c8ra08915a
- Seifert MHJ (2005) ProPose: steered virtual screening by simultaneous protein-ligand docking and ligand-ligand alignment. J Chem Inf Model. https://doi.org/10.1021/ci0496393
- Schellhammer I, Rarey M (2007) TrixX: Structure-based molecule indexing for large-scale virtual screening in sublinear time. J Comput Aided Mol Des. https://doi.org/10.1007/s10822-007-9103-5
- Lagarde N, Goldwaser E, Pencheva T et al (2019) A free webbased protocol to assist structure-based virtual screening experiments. Int J Mol Sci. https://doi.org/10.3390/ijms20184648
- Rifaioglu AS, Nalbat E, Atalay V et al (2020) DEEPScreen: high performance drug-target interaction prediction with convolutional neural networks using 2-D structural compound representations. Chem Sci. https://doi.org/10.1039/c9sc03414e
- Obrezanova O, Segall MD (2010) Gaussian processes for classification: QSAR modeling of ADMET and target activity. J Chem Inf Model. https://doi.org/10.1021/ci900406x
- Wu Z, Zhu M, Kang Y et al (2020) Do we need different machine learning algorithms for QSAR modeling? A comprehensive assessment of 16 machine learning algorithms on 14 QSAR data sets. Brief Bioinform. https://doi.org/10.1093/bib/bbaa321
- Obrezanova O, Csányi G, Gola JMR, Segall MD (2007) Gaussian processes: a method for automatic QSAR modeling of ADME properties. J Chem Inf Model. https://doi.org/10.1021/ci7000633
- Benfenati E, Manganaro A, Gini G (2013) VEGA-QSAR: AI inside a platform for predictive toxicology. In: CEUR Workshop Proceedings
- Ambure P, Halder AK, González Díaz H, Cordeiro MNDS (2019) QSAR-Co: an open source software for developing robust multitasking or multitarget classification-based QSAR models. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.9b00295
- Chen S, Xue D, Chuai G et al (2020) FL-QSAR: a federated learning based QSAR prototype for collaborative drug discovery. Bioinformatics. https://doi.org/10.1093/bioinformatics/btaa1006
- Olier I, Sadawi N, Bickerton GR et al (2018) Meta-QSAR: a large-scale application of meta-learning to drug design and discovery. Mach Learn. https://doi.org/10.1007/s10994-017-5685-x
- Soufan O, Ba-Alawi W, Magana-Mora A et al (2018) DPubChem: a web tool for QSAR modeling and high-throughput virtual screening. Sci Rep. https://doi.org/10.1038/s41598-018-27495-x
- Karpov P, Godin G, Tetko IV (2020) Transformer-CNN: swiss knife for QSAR modeling and interpretation. J Cheminform. https://doi.org/10.1186/s13321-020-00423-w
- Wang Y-L, Wang F, Shi X-X et al (2020) Cloud 3D-QSAR: a web tool for the development of quantitative structure–activity relationship models in drug discovery. Brief Bioinform. https://doi.org/10.1093/bib/bbaa276
- Goh GB, Siegel C, Vishnu A, et al (2017) Chemception: A deep neural network with minimal chemistry knowledge matches the performance of expert-developed QSAR/QSPR models. arXiv
- Reis J, Cagide F, Chavarria D et al (2016) Discovery of new chemical entities for old targets: insights on the lead optimization of chromone-based monoamine oxidase B (MAO-B) inhibitors. J Med Chem. https://doi.org/10.1021/acs.jmedchem.6b00527
- Hoelz L, Horta B, Araújo J et al (2010) Quantitative structureactivity relationships of antioxidant phenolic compounds. J Chem Pharm Res 2(5):291–306
- Zhang Y, Han Z, Gao Q et al (2019) Prediction of K562 Cells Functional Inhibitors Based on Machine Learning Approaches. Curr Pharm Des. https://doi.org/10.2174/1381612825666191107092214
- Halder AK, Giri AK, Dias Soeiro Cordeiro MN (2019) Multitarget chemometric modelling, fragment analysis and virtual screening with ERK inhibitors as potential anticancer agents. Molecules. https://doi.org/10.3390/molecules24213909
- Halder AK, Cordeiro MNDS (2019) Development of multitarget chemometric models for the inhibition of class I PI3K enzyme isoforms: a case study using QSAR-Co tool. Int J Mol Sci. https://doi.org/10.3390/ijms20174191
- Kim S, Cho KH (2019) PyQSAR: a fast QSAR modeling platform using machine learning and jupyter notebook. Bull Korean Chem Soc. https://doi.org/10.1002/bkcs.11638
- Ben Geoffrey AS, Christian Prasana J, Muthu S (2020) Structureactivity relationship of Quercetin and its tumor necrosis factor alpha inhibition activity by computational and machine learning methods. Mater Today Proc. https://doi.org/10.1016/j.matpr. 2020.07.464
- Ben Geoffrey A S, Rafal Madaj, Akhil Sanker, Mario Sergio Valdés Tresanco, Host Antony Davidd, Gitanjali Roy, Rinnu Sarah Saji, Abdulbasit Haliru Yakubu BM Automated In Silico Identification of Drug Candidates for Coronavirus Through a Novel Programmatic Tool and Extensive Computational (MD, DFT) Studies of Select Drug Candidatesl; https://doi.org/https://doi.org/10.26434/chemrxiv.12423638.v3
- Žuvela P, David J, Wong MW (2018) Interpretation of ANNbased QSAR models for prediction of antioxidant activity of flavonoids. J Comput Chem. https://doi.org/10.1002/jcc.25168
- Ding Q, Hou S, Zu S et al (2020) VISAR: an interactive tool for dissecting chemical features learned by deep neural network QSAR models. Bioinformatics. https://doi.org/10.1093/bioinformatics/btaa187
- Gadaleta D, Manganelli S, Roncaglioni A et al (2018) QSAR modeling of ToxCast assays relevant to the molecular initiating events of AOPs leading to hepatic steatosis. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.8b00297
- Hermansyah O, Bustamam A, Yanuar A (2020) Virtual Screening of DPP-4 Inhibitors Using QSAR-Based Artificial Intelligence and Molecular Docking of Hit Compounds to DPP-8 and DPP-9 Enzymes. https://doi.org/10.21203/rs.2.22282/v1
- Tian Y, Zhang S, Yin H, Yan A (2020) Quantitative structureactivity relationship (QSAR) models and their applicability domain analysis on HIV-1 protease inhibitors by machine learning methods. Chemom Intell Lab Syst. https://doi.org/10.1016/j.chemolab.2019.103888
- Wei Y, Li W, Du T et al (2019) Targeting HIV/HCV coinfection using a machine learning-based multiple quantitative structureActivity Relationships (Multiple QSAR) Method. Int J Mol Sci. https://doi.org/10.3390/ijms20143572
- Michel Kana (2020) Handling Missing Data For Advanced Machine Learning
- Kumar S (2020) 7 Ways to Handle Missing Values in Machine Learning | by Satyam Kumar | Towards Data Science
- Gad SC (2014) QSAR. In: Third E (ed) Wexler PBT- Encyclopedia of Toxicology. Academic Press, Oxford, pp 1–9
- Neves BJ, Braga RC, Melo-Filho CC et al (2018) QSAR-Based Virtual Screening: Advances and Applications in Drug Discovery. Front Pharmacol 9:1275. https://doi.org/10.3389/fphar.2018.01275
- Roy K, Kar S, Das RN (2015) Chapter 9 – Newer QSAR Techniques. In: Roy K, Kar S, Das RN, Book Title- Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment (eds). Academic Press, Boston,
- Kwon S, Bae H, Jo J, Yoon S (2019) Comprehensive ensemble in QSAR prediction for drug discovery. BMC Bioinformatics 20:521. https://doi.org/10.1186/s12859-019-3135-4
- Roy K, Kar S, Das RN (2015) Chapter 12 – Future Avenues. In: Roy K, Kar S, Das RN, Book Title- Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment (eds). Academic Press, Boston, pp 455–462. https://doi.org/https://doi.org/10.1016/B978-0-12-801505-6.00012-0
- Paolini GV, Shapland RHB, Van Hoorn WP et al (2006) Global mapping of pharmacological space. Nat Biotechnol. https://doi.org/10.1038/nbt1228
- Koch U, Hamacher M, Nussbaumer P (2014) Cheminformatics at the interface of medicinal chemistry and proteomics. Biochim Biophys Acta-Proteins Proteomics 1844(1):156–61; https://doi.org/10.1016/j.bbapap.2013.05.010
- Makhouri FR, Ghasemi JB (2019) Combating diseases with computational strategies used for drug design and discovery. Curr Top Med Chem. https://doi.org/10.2174/1568026619666190121125106
- Würth R, Thellung S, Bajetto A et al (2016) Drug-repositioning opportunities for cancer therapy: novel molecular targets for known compounds. Drug Discov Today 21(1):190–199. https://doi.org/10.1016/j.drudis.2015.09.017
- Joachim Haupt V, Schroeder M (2011) Old friends in new guise: repositioning of known drugs with structural bioinformatics. Brief Bioinform. https://doi.org/10.1093/bib/bbr011
- Butcher EC (2005) Can cell systems biology rescue drug discovery? Nat Rev Drug Discov. https://doi.org/10.1038/nrd1754
- Iyengar R, Zhao S, Chung SW et al (2012) Merging systems biology with pharmacodynamics. Sci Transl Med 4(126):126ps7. https://doi.org/10.1126/scitranslmed.3003563
- Martínez V, Navarro C, Cano C et al (2015) DrugNet: networkbased drug-disease prioritization by integrating heterogeneous data. Artif Intell Med. https://doi.org/10.1016/j.artmed.2014.11.003
- Zhang W, Xu H, Li X et al (2020) DRIMC: an improved drug repositioning approach using Bayesian inductive matrix completion. Bioinformatics. https://doi.org/10.1093/bioinformatics/btaa062
- Luo H, Zhang P, Cao XH et al (2016) DPDR-CPI, a server that predicts drug positioning and drug repositioning via chemical-protein interactome. Sci Rep. https://doi.org/10.1038/srep35996
- Zhu Q, Tao C, Shen F, Chute CG (2014) Exploring the pharmacogenomics knowledge base (pharmgkb) for repositioning breast cancer drugs by leveraging Web ontology language (owl) and cheminformatics approaches. In: Pacific Symposium on Biocomputing
- Gallo K, Goede A, Eckert A et al (2020) PROMISCUOUS 2.0: a resource for drug-repositioning. Nucleic Acids Res. https://doi.org/10.1093/nar/gkaa1061
- Luo H, Li M, Wang S et al (2018) Computational drug repositioning using low-rank matrix approximation and randomized algorithms. Bioinformatics. https://doi.org/10.1093/bioinformatics/bty013
- Yella JK, Jegga AG (2020) MGATRx: discovering drug repositioning candidates using multi-view graph attention. biorxiv. https://doi.org/10.1101/2020.06.29.171876
- Yan CK, Wang WX, Zhang G et al (2019) BiRWDDA: a novel drug repositioning method based on multisimilarity fusion. J Comput Biol. https://doi.org/10.1089/cmb.2019.0063
- Fahimian G, Zahiri J, Arab SS, Sajedi RH (2019) RepCOOL: computational drug repositioning via integrating heterogeneous biological networks. biorxiv. https://doi.org/10.1101/817882
- Li Z, Yao Y, Cheng X, et al (2020) A Computational Framework of Host-Based Drug Repositioning for Broad-Spectrum Antivirals against RNA Viruses. https://doi.org/10.26434/chemrxiv.12927260.v1
- Wu D, Gao W, Li X et al (2020) Dr AFC: drug repositioning through anti-fibrosis characteristic. Brief Bioinform. https://doi. org/10.1093/bib/bbaa115
- Hooshmand SA, Zarei Ghobadi M, Hooshmand SE et al (2020) A multimodal deep learning-based drug repurposing approach for treatment of COVID-19. Mol Divers. https://doi.org/10.1007/s11030-020-10144-9
- Zhou Y, Hou Y, Shen J et al (2020) Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. https://doi.org/10.1038/s41421-020-0153-3
- Zheng X, He S, Song X, et al (2018) DTI-RCNN: New efficient hybrid neural network model to predict drug–target interactions. In: Lecture Notes in Computer Science (including subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics)
- Jarada TN, Rokne JG, Alhajj R (2020) SNF–CVAE: computational method to predict drug–disease interactions using similarity network fusion and collective variational autoencoder. Knowledge-Based Syst. https://doi.org/10.1016/j.knosys.2020.106585
- Xu R, Wang QQ (2015) PhenoPredict: a disease phenome-wide drug repositioning approach towards schizophrenia drug discovery. J Biomed Inform. https://doi.org/10.1016/j.jbi.2015.06.027
- Wu Z, Cheng F, Li J et al (2017) SDTNBI: an integrated network and chemoinformatics tool for systematic prediction of drug-target interactions and drug repositioning. Brief Bioinform. https://doi.org/10.1093/bib/bbw012
- Zeng X, Zhu S, Liu X et al (2019) DeepDR: a network-based deep learning approach to in silico drug repositioning. Bioinformatics. https://doi.org/10.1093/bioinformatics/btz418
- Chen H, Cheng F, Li J (2020) IDrug: Integration of drug repositioning and drug-target prediction via cross-network embedding. PLoS Comput Biol. https://doi.org/10.1371/journal.pcbi.1008040
- Li B, Dai C, Wang L et al (2020) A novel drug repurposing approach for non-small cell lung cancer using deep learning. PLoS ONE. https://doi.org/10.1371/journal.pone.0233112
- Kuenzi BM, Park J, Fong SH et al (2020) Predicting drug response and synergy using a deep learning model of human cancer cells. Cancer Cell. https://doi.org/10.1016/j.ccell.2020.09.014
- Wang Z, Zhou M, Arnold C (2020) Toward heterogeneous information fusion: bipartite graph convolutional networks for in silico drug repurposing. Bioinformatics. https://doi.org/10.1093/bioinformatics/btaa437
- Pinzi L, Rastelli G (2019) Molecular docking: Shifting paradigms in drug discovery. Int J Mol Sci. https://doi.org/10.3390/ijms20184331
- Muhammed MT, Aki-Yalcin E (2019) Homology modeling in drug discovery: overview, current applications, and future perspectives. Chem Biol Drug Des 93:12–20. https://doi.org/10.1111/cbdd.13388
- Lynch SR, Bothwell T, Campbell L et al (2007) A comparison of physical properties, screening procedures and a human efficacy trial for predicting the bioavailability of commercial elemental iron powders used for food fortification. Int J Vitam Nutr Res. https://doi.org/10.1024/0300-9831.77.2.107
- Schneider P, Walters WP, Plowright AT et al (2020) Rethinking drug design in the artificial intelligence era. Nat Rev Drug Discov 19:353–364. https://doi.org/10.1038/s41573-019-0050-3
- Chen H, Engkvist O, Wang Y et al (2018) The rise of deep learning in drug discovery. Drug Discov Today 23(6):1241–1250. https://doi.org/10.1016/j.drudis.2018.01.039
- Lusci A, Pollastri G, Baldi P (2013) Deep architectures and deep learning in chemoinformatics: the prediction of aqueous solubility for drug-like molecules. J Chem Inf Model. https://doi.org/10.1021/ci400187y
- Kumar R, Sharma A, Siddiqui MH, Tiwari RK (2017) Prediction of human intestinal absorption of compounds using artificial intelligence techniques. Curr Drug Discov Technol. https://doi.org/10.2174/1570163814666170404160911
- Zang Q, Mansouri K, Williams AJ et al (2017) In silico prediction of physicochemical properties of environmental chemicals using molecular fingerprints and machine learning. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.6b00625
- Tetko IV, Gasteiger J, Todeschini R et al (2005) Virtual computational chemistry laboratory-design and description. J Comput Aided Mol Des. https://doi.org/10.1007/s10822-005-8694-y
- Radchenko E V, Palyulin VA, Zefirov NS (2002) Virtual computational chemistry laboratory. System
- Royal Society of Chemistry (2015) ChemSpider. Search and Share Chemistry. R. Soc, Chem
- Kucukdereli H, Allen NJ, Lee AT et al (2011) Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins hevin and SPARC. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.1104977108
- Ayati A, Falahati M, Irannejad H, Emami S (2012) Synthesis, in vitro antifungal evaluation and in silico study of 3-azolyl-4-chromanone phenylhydrazones. DARU, J Pharm Sci. https://doi.org/10.1186/2008-2231-20-46
- Rashid M (2020) Design, synthesis and ADMET prediction of bis-benzimidazole as anticancer agent. Bioorg Chem. https://doi.org/10.1016/j.bioorg.2020.103576
- Puratchikody A, Sriram D, Umamaheswari A, Irfan N (2016) 3-D structural interactions and quantitative structural toxicity studies of tyrosine derivatives intended for safe potent inflammation treatment. Chem Cent J. https://doi.org/10.1186/s13065-016-0169-9
- Nascimento ACA, Prudêncio RBC, Costa IG (2016) A multiple kernel learning algorithm for drug-target interaction prediction. BMC Bioinformatics. https://doi.org/10.1186/s12859-016-0890-3
- Öztürk H, Özgür A, Ozkirimli E (2018) A chemical language based approach for protein-Ligand interaction prediction. arXiv https://doi.org/10.1002/minf.202000212
- Nascimento ACA, Prudêncio RBC, Costa IG (2019) A drugtarget network-based supervised machine learning repurposing method allowing the use of multiple heterogeneous information sources. Methods Mol Biol 1903:281–289. https://doi.org/10.1007/978-1-4939-8955-3_17
- Öztürk H, Özgür A, Ozkirimli E (2018) DeepDTA: Deep drugtarget binding affinity prediction. Bioinformatics 34(17):i821–i829. https://doi.org/10.1093/bioinformatics/bty593
- Feng Q, Dueva E, Cherkasov A, Ester M (2018) PADME: A deep learning-based framework for drug-target interaction prediction. arXiv https://arxiv.org/abs/1807.09741v4
- Beck BR, Shin B, Choi Y et al (2020) Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput Struct Biotechnol J. https://doi.org/10.1016/j.csbj.2020.03.025
- Lee H, Kim W (2019) Comparison of target features for predicting drug-target interactions by deep neural network based on large-scale drug-induced transcriptome data. Pharmaceutics. https://doi.org/10.3390/pharmaceutics11080377
- Karimi M, Wu D, Wang Z, Shen Y (2019) DeepAffinity: interpretable deep learning of compound-protein affinity through unified recurrent and convolutional neural networks. Bioinformatics. https://doi.org/10.1093/bioinformatics/btz111
- Born J, Manica M, Cadow J, et al (2020) PaccMannRL on SARS-CoV-2: Designing antiviral candidates with conditional generative models. arXiv https://arxiv.org/abs/2005.13285v3
- Jiang M, Li Z, Bian Y, Wei Z (2019) A novel protein descriptor for the prediction of drug binding sites. BMC Bioinformatics. https://doi.org/10.1186/s12859-019-3058-0
- Cañada A, Capella-Gutierrez S, Rabal O et al (2017) LimTox: a web tool for applied text mining of adverse event and toxicity associations of compounds, drugs and genes. Nucleic Acids Res. https://doi.org/10.1093/nar/gkx462
- Pires DEV, Blundell TL, Ascher DB (2015) pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem. https://doi.org/10.1021/acs.jmedchem.5b00104
- Cheng F, Li W, Zhou Y et al (2012) AdmetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model. https://doi.org/10.1021/ci300367a
- Patlewicz G, Jeliazkova N, Safford RJ et al (2008) An evaluation of the implementation of the Cramer classification scheme in the Toxtree software. SAR QSAR Environ Res. https://doi.org/10.1080/10629360802083871
- Uygun MT, Amudi K, Turaçlı İD, Menges N (2021) A new synthetic approach for pyrazolo[1,5-a]pyrazine-4(5H)-one derivatives and their antiproliferative effects on lung adenocarcinoma cell line. Mol Divers. https://doi.org/10.1007/s11030-020-10161-8
- Srivastava A, Siddiqui S, Ahmad R et al (2020) Exploring nature’s bounty: identification of Withania somnifera as a promising source of therapeutic agents against COVID-19 by virtual screening and in silico evaluation. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1835725
- Attene-Ramos MS, Miller N, Huang R et al (2013) The Tox21 robotic platform for the assessment of environmental chemicalsFrom vision to reality. Drug Discov. Today 18(15–16):716–23. https://doi.org/10.1016/j.drudis.2013.05.015
- Wang Z, Liang L, Yin Z, Lin J (2016) Improving chemical similarity ensemble approach in target prediction. J Cheminform. https://doi.org/10.1186/s13321-016-0130-x
- Pu L, Naderi M, Liu T et al (2019) eToxPred: a machine learningbased approach to estimate the toxicity of drug candidates. BMC Pharmacol Toxicol. https://doi.org/10.1186/s40360-018-0282-6
- Lysenko A, Sharma A, Boroevich KA, Tsunoda T (2018) An integrative machine learning approach for prediction of toxicityrelated drug safety. Life Sci Alliance. https://doi.org/10.26508/lsa.201800098
- Zhou B, Sun Q, Kong DX (2016) Predicting cancer-relevant proteins using an improved molecular similarity ensemble approach. Oncotarget. https://doi.org/10.18632/oncotarget.8716
- Huang R, Xia M, Sakamuru S et al (2016) Modelling the Tox21 10 K chemical profiles for in vivo toxicity prediction and mechanism characterization. Nat Commun. https://doi.org/10.1038/ncomms10425
- Gupta VK, Rana PS (2019) Toxicity prediction of small drug molecules of androgen receptor using multilevel ensemble model. J Bioinform Comput Biol. https://doi.org/10.1142/S0219720019500331
- Mayr A, Klambauer G, Unterthiner T, Hochreiter S (2016) DeepTox: toxicity prediction using deep learning. Front Environ Sci. https://doi.org/10.3389/fenvs.2015.00080
- Gayvert KM, Madhukar NS, Elemento O (2016) A data-driven approach to predicting successes and failures of clinical trials. Cell Chem Biol. https://doi.org/10.1016/j.chembiol.2016.07.023
- Gilvary C, Elkhader J, Madhukar N et al (2020) A machine learning and network framework to discover new indications for small molecules. PLoS Comput Biol. https://doi.org/10.1371/JOURNAL.PCBI.1008098
- Robledo-Cadena DX, Gallardo-Pérez JC, Dávila-Borja V et al (2020) Non-steroidal anti-inflammatory drugs increase cisplatin, paclitaxel, and doxorubicin efficacy against human cervix cancer cells. Pharmaceuticals (Basel). https://doi.org/10.3390/ph13120463
- Simm J, Klambauer G, Arany A et al (2018) Repurposing highthroughput image assays enables biological activity prediction for drug discovery. Cell Chem Biol. https://doi.org/10.1016/j.chembiol.2018.01.015
- Goh GB, Siegel C, Hodas N, Vishnu A (2017) SMILES2vec: An interpretable general-purpose deep neural network for predicting chemical properties. arXiv https://arxiv.org/abs/1712.02034v2
- Preuer K, Lewis RPI, Hochreiter S et al (2018) Deepsynergy: predicting anti-cancer drug synergy with deep learning. Bioinformatics. https://doi.org/10.1093/bioinformatics/btx806
- Xu Y, Pei J, Lai L (2017) Deep learning based regression and multiclass models for acute oral toxicity prediction with automatic chemical feature extraction. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.7b00244
- Rodrigues T, Werner M, Roth J et al (2018) Machine intelligence decrypts β-lapachone as an allosteric 5-lipoxygenase inhibitor. Chem Sci. https://doi.org/10.1039/c8sc02634c
- Luechtefeld T, Marsh D, Rowlands C, Hartung T (2018) Machine learning of toxicological big data enables read-across structure activity relationships (RASAR) outperforming animal test reproducibility. Toxicol Sci. https://doi.org/10.1093/toxsci/kfy152
- Zhang C, Cheng F, Li W et al (2016) In silico prediction of drug induced liver toxicity using substructure pattern recognition method. Mol Inform. https://doi.org/10.1002/minf.201500055
- Lei T, Li Y, Song Y et al (2016) ADMET evaluation in drug discovery: 15. Accurate prediction of rat oral acute toxicity using relevance vector machine and consensus modeling. J Cheminform. https://doi.org/10.1186/s13321-016-0117-7
- Lei T, Chen F, Liu H et al (2017) ADMET evaluation in drug discovery. Part 17: development of quantitative and qualitative prediction models for chemical-induced respiratory toxicity. Mol Pharm. https://doi.org/10.1021/acs.molpharmaceut.7b00317
- Lei T, Sun H, Kang Y et al (2017) ADMET evaluation in drug discovery. 18 reliable prediction of chemical-induced urinary tract toxicity by boosting machine learning approaches. Mol Pharm. https://doi.org/10.1021/acs.molpharmaceut.7b00631
- Pandya R, Pandya J (2015) C5.0 algorithm to improved decision tree with feature selection and reduced error pruning. Int J Comput Appl. https://doi.org/10.5120/20639-3318
- Jenwitheesuk E, Horst JA, Rivas KL et al (2008) Novel paradigms for drug discovery: computational multitarget screening. Trends Pharmacol Sci. https://doi.org/10.1016/j.tips.2007.11.007
- Gu S, Lai L, hua, (2020) Associating 197 Chinese herbal medicine with drug targets and diseases using the similarity ensemble approach. Acta Pharmacol Sin 41:432–438. https://doi.org/10.1038/s41401-019-0306-9
- Chen YT, Xie JY, Sun Q, Mo WJ (2019) Novel drug candidates for treating esophageal carcinoma: a study on differentially expressed genes, using connectivity mapping and molecular docking. Int J Oncol 54:152–166. https://doi.org/10.3892/ijo.2018.4618
- Taha KF, Khalil M, Abubakr MS, Shawky E (2020) Identifying cancerrelated molecular targets of Nandina domestica Thunb. by network pharmacologybased analysis in combination with chemical profiling and molecular docking studies. J Ethnopharmacol 249:112413. https://doi.org/10.1016/j.jep.2019.112413
- Anighoro A, Bajorath J, Rastelli G (2014) Polypharmacology: Challenges and opportunities in drug discovery. J Med Chem 57(19):7874–87. https://doi.org/10.1021/jm5006463
- Zhang W, Pei J, Lai L (2017) Computational multitarget drug design. J Chem Inf Model 57(3):403–412. https://doi.org/10.1021/acs.jcim.6b00491
- Proschak E, Stark H, Merk D (2019) Polypharmacology by design: a medicinal chemist’s perspective on multitargeting compounds. J Med Chem 62(2):420–444. https://doi.org/10.1021/acs.jmedchem.8b00760
- Awale M, Reymond JL (2017) The polypharmacology browser: a web-based multi-fingerprint target prediction tool using ChEMBL bioactivity data. J Cheminform. https://doi.org/10.1186/s13321-017-0199-x
- Reker D, Rodrigues T, Schneider P, Schneider G (2014) Identifying the macromolecular targets of de novo-designed chemical entities through self-organizing map consensus. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.1320001111
- Wang L, Ma C, Wipf P et al (2013) Targethunter: an in silico target identification tool for predicting therapeutic potential of small organic molecules based on chemogenomic database. AAPS J. https://doi.org/10.1208/s12248-012-9449-z
- Xia W, Chenxu P, Honglin L (2016) PharmMapper. In: Enhancing Enrich. Pharmacophore-Based Target Predict. Polypharmacological Profiles Drugs 56(6):1175–83. https://doi.org/10.1021/acs.jcim.5b00690
- Gong J, Cai C, Liu X et al (2013) ChemMapper: a versatile web server for exploring pharmacology and chemical structure association based on molecular 3D similarity method. Bioinformatics. https://doi.org/10.1093/bioinformatics/btt270
- Gfeller D, Grosdidier A, Wirth M et al (2014) SwissTargetPrediction: a web server for target prediction of bioactive small molecules. Nucleic Acids Res. https://doi.org/10.1093/nar/gku293
- Poirier M, Awale M, Roelli MA et al (2019) Identifying lysophosphatidic acid acyltransferase β (LPAAT-β) as the target of a nanomolar angiogenesis inhibitor from a phenotypic screen using the polypharmacology browser PPB2. ChemMedChem. https://doi.org/10.1002/cmdc.201800554
- Ozhathil LC, Delalande C, Bianchi B et al (2018) Identification of potent and selective small molecule inhibitors of the cation channel TRPM4. Br J Pharmacol. https://doi.org/10.1111/bph.14220
- Ratnawati DE, Marjono M, Anam S (2018) Prediction of active compounds from SMILES codes using backpropagation algorithm. In: AIP Conference Proceedings
- Van Vleet TR, Liguori MJ, Lynch JJ et al (2019) Screening strategies and methods for better offtarget liability prediction and identification of small-molecule pharmaceuticals. SLAS Discov 24(1):1–24. https://doi.org/10.1177/2472555218799713
- Yue SJ, Liu J, Feng WW et al (2017) System pharmacologybased dissection of the synergistic mechanism of huangqi and huanglian for diabetes mellitus. Front Pharmacol. https://doi.org/10.3389/fphar.2017.00694
- Shi XQ, Yue SJ, Tang YP et al (2019) A network pharmacology approach to investigate the blood enriching mechanism of Danggui buxue Decoction. J Ethnopharmacol. https://doi.org/10.1016/j.jep.2019.01.027
- Liu X, Wu J, Zhang D et al (2018) A network pharmacology approach to uncover the multiple mechanisms of hedyotis diffusa willd on colorectal cancer. EvidenceBased Complem Altern Med. https://doi.org/10.1155/2018/6517034
- Wang J, Luo C, Shan C et al (2015) Inhibition of human copper trafficking by a small molecule significantly attenuates cancer cell proliferation. Nat Chem. https://doi.org/10.1038/nchem.2381
- Fang J, Li Y, Liu R et al (2015) Discovery of multitarget-directed ligands against Alzheimer’s disease through systematic prediction of chemical-protein interactions. J Chem Inf Model. https://doi.org/10.1021/ci500574n
- Gao L, Wang KX, Zhou YZ et al (2018) Uncovering the anticancer mechanism of compound Kushen Injection against HCC by integrating quantitative analysis, network analysis and experimental validation. Sci Rep. https://doi.org/10.1038/s41598-017-18325-7
- Zhou W, Liu X, Tu Z et al (2013) Discovery of pteridin-7(8H)-one-based irreversible inhibitors targeting the epidermal growth factor receptor (EGFR) kinase T790M/L858R Mutant. J Med Chem 56:7821–7837. https://doi.org/10.1021/jm401045n
- Wang Q, Feng YH, Huang JC et al (2017) A novel framework for the identification of drug target proteins: combining stacked auto-encoders with a biased support vector machine. PLoS ONE. https://doi.org/10.1371/journal.pone.0176486
- Carvalho-Silva D, Pierleoni A, Pignatelli M et al (2019) Open targets platform: new developments and updates two years on. Nucleic Acids Res. https://doi.org/10.1093/nar/gky1133
- López-Cortés A, Paz-y-Miño C, Guerrero S et al (2020) Pharmacogenomics, biomarker network, and allele frequencies in colorectal cancer. Pharmacogenomics J 20(1):136–158. https://doi.org/10.1038/s41397-019-0102-4
- Nabirotchkin S, Peluffo AE, Bouaziz J, Cohen D (2020) Focusing on the unfolded protein response and autophagy related pathways to reposition common approved drugs against COVID-19. Preprints
- López-Isac E, Acosta-Herrera M, Kerick M et al (2019) GWAS for systemic sclerosis identifies multiple risk loci and highlights fibrotic and vasculopathy pathways. Nat Commun. https://doi.org/10.1038/s41467-019-12760-y
- Martin P, Ding J, Duffus K et al (2019) Chromatin interactions reveal novel gene targets for drug repositioning in rheumatic diseases. Ann Rheum Dis. https://doi.org/10.1136/annrheumdis-2018-214649
- Dong J, Cao DS, Miao HY et al (2015) ChemDes: an integrated web-based platform for molecular descriptor and fingerprint computation. J Cheminform. https://doi.org/10.1186/s13321-015-0109-z
- Angelo RM, Io AK, Almeida MP, et al (2020) OntoQSAR: An ontology for interpreting chemical and biological data in quantitative structure-activity relationship studies. In: Proceedings-14th IEEE International Conference on Semantic Computing, ICSC 2020
- Oldenhof M, Arany A, Moreau Y, Simm J (2020) Chemgrapher: optical graph recognition of chemical compounds by deep learning. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.0c00459
- Dong J, Yao ZJ, Zhu MF et al (2017) ChemSAR: An online pipelining platform for molecular SAR modeling. J Cheminform. https://doi.org/10.1186/s13321-017-0215-1
- Buyukbingol E, Sisman A, Akyildiz M et al (2007) Adaptive neuro-fuzzy inference system (ANFIS): a new approach to predictive modeling in QSAR applications: a study of neuro-fuzzy modeling of PCP-based NMDA receptor antagonists. Bioorg Med Chem 15:4265–4282. https://doi.org/10.1016/j.bmc.2007.03.065
- Jiang HJ, Huang YA, You ZH (2019) Predicting drug-disease associations via using Gaussian interaction profile and kernelbased autoencoder. Biomed Res Int. https://doi.org/10.1155/2019/2426958
- Wang YY, Cui C, Qi L et al (2019) DrPOCS: drug repositioning based on projection onto convex sets. IEEE/ACM Trans Comput Biol Bioinforma. https://doi.org/10.1109/TCBB.2018.2830384
- Xuan P, Cui H, Shen T et al (2019) HeteroDualNet: a dual convolutional neural network with heterogeneous layers for drugdisease association prediction via chou’s five-step rule. Front Pharmacol. https://doi.org/10.3389/fphar.2019.01301
- Sadeghi SS, Keyvanpour M (2019) RCDR: A Recommender Based Method for Computational Drug Repurposing. In: 2019 IEEE 5th Conference on Knowledge Based Engineering and Innovation, KBEI 2019
- Zhu Q, Luo J, Ding P, Xiao Q (2018) GRTR: Drug-disease association prediction based on graph regularized transductive regression on heterogeneous network. In: Lecture Notes in Computer Science (including subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics)
- Jiang HJ, Huang YA, You ZH (2020) SAEROF: an ensemble approach for large-scale drug-disease association prediction by incorporating rotation forest and sparse autoencoder deep neural network. Sci Rep. https://doi.org/10.1038/s41598-020-61616-9
- Wang MN, You ZH, Li LP, et al (2020) WGMFDDA: A Novel Weighted-Based Graph Regularized Matrix Factorization for Predicting Drug-Disease Associations. In: Lecture Notes in Computer Science (including subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics),
- Liu H, Zhang W, Song Y et al (2020) HNet-DNN: inferring new drug-disease associations with deep neural network based on heterogeneous network features. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.9b01008
- Lee I, Keum J, Nam H (2019) DeepConv-DTI: prediction of drug-target interactions via deep learning with convolution on protein sequences. PLoS Comput Biol. https://doi.org/10.1371/journal.pcbi.1007129
- Abdel-Basset M, Hawash H, Elhoseny M et al (2020) DeepHDTA: deep learning for predicting drug-target interactions: a case study of COVID-19 drug repurposing. IEEE Access. https://doi.org/10.1109/access.2020.3024238
- Yang J, He S, Zhang Z, Bo X (2020) NegStacking: drug-target interaction prediction based on ensemble learning and logistic regression. IEEE/ACM Trans Comput Biol Bioinforma. https://doi.org/10.1109/TCBB.2020.2968025
- King MD, Long T, Pfalmer DL et al (2018) SPIDR: small-molecule peptide-influenced drug repurposing. BMC Bioinformatics. https://doi.org/10.1186/s12859-018-2153-y
- Huang K, Fu T, Glass LM et al (2020) DeepPurpose: a deep learning library for drug–target interaction prediction. Bioinformatics. https://doi.org/10.1093/bioinformatics/btaa1005
- Chu Y, Kaushik AC, Wang X et al (2019) DTI-CDF: a cascade deep forest model towards the prediction of drug-target interactions based on hybrid features. Brief Bioinform. https://doi.org/10.1093/bib/bbz152
- Shar PA, Tao W, Gao S et al (2016) Pred-binding: large-scale protein–ligand binding affinity prediction. J Enzyme Inhib Med Chem 31:1443–1450. https://doi.org/10.3109/14756366.2016.1144594
- Capuzzi SJ, Kim ISJ, Lam WI et al (2017) Chembench: a publicly accessible, integrated cheminformatics portal. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.6b00462
- Pires DEV, Blundell TL, Ascher DB (2016) MCSM-lig: quantifying the effects of mutations on protein-small molecule affinity in genetic disease and emergence of drug resistance. Sci Rep. https://doi.org/10.1038/srep29575
- Pires DEV, Ascher DB (2016) CSM-lig: a web server for assessing and comparing protein-small molecule affinities. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw390
- Pires DEV, Ascher DB (2016) mCSM-AB: a web server for predicting antibody-antigen affinity changes upon mutation with graph-based signatures. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw458
- Kaminskas LM, Pires DEV, Ascher DB (2019) dendPoint: a web resource for dendrimer pharmacokinetics investigation and prediction. Sci Rep. https://doi.org/10.1038/s41598-019-51789-3
- Patel RD, Prasanth Kumar S, Pandya HA, Solanki HA (2018) MDCKpred: a web-tool to calculate MDCK permeability coefficient of small molecule using membrane-interaction chemical features. Toxicol Mech Methods. https://doi.org/10.1080/15376516.2018.1499840
- Montanari F, Knasmüller B, Kohlbacher S et al (2020) Vienna LiverTox workspace—a set of machine learning models for prediction of interactions profiles of small molecules with transporters relevant for regulatory agencies. Front Chem. https://doi.org/10.3389/fchem.2019.00899
- Kochev N, Avramova S, Jeliazkova N (2018) Ambit-SMIRKS: a software module for reaction representation, reaction search and structure transformation. J Cheminform. https://doi.org/10.1186/s13321-018-0295-6
- Hornig M, Klamt A (2005) COSMOfrag: a novel tool for highthroughput ADME property prediction and similarity screening based on quantum chemistry. J Chem Inf Model. https://doi.org/10.1021/ci0501948
- Hassan-Harrirou H, Zhang C, Lemmin T (2020) RosENet: improving binding affinity prediction by leveraging molecular mechanics energies with an ensemble of 3D convolutional neural networks. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.0c00075
- Rifaioglu AS, Cetin Atalay R, Cansen Kahraman D et al (2020) MDeePred: novel multi-channel protein featurization for deep learning-based binding affinity prediction in drug discovery. Bioinformatics. https://doi.org/10.1093/bioinformatics/btaa858
- Banerjee P, Eckert AO, Schrey AK, Preissner R (2018) ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. https://doi.org/10.1093/nar/gky318
- Dong J, Wang NN, Yao ZJ et al (2018) Admetlab: a platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. J Cheminform. https://doi.org/10.1186/s13321-018-0283-x
- Maunz A, Gütlein M, Rautenberg M et al (2013) Lazar: a modular predictive toxicology framework. Front Pharmacol. https://doi.org/10.3389/fphar.2013.00038
- Yao ZJ, Dong J, Che YJ et al (2016) TargetNet: a web service for predicting potential drug–target interaction profiling via multitarget SAR models. J Comput Aided Mol Des. https://doi.org/10.1007/s10822-016-9915-2
- Meng C, Hu Y, Zhang Y, Guo F (2020) PSBP-SVM: a machine learning-based computational identifier for predicting polystyrene binding peptides. Front Bioeng Biotechnol. https://doi.org/10.3389/fbioe.2020.00245
- Shen C, Luo J, Ouyang W et al (2020) IDDkin: network-based influence deep diffusion model for enhancing prediction of kinase inhibitors. Bioinformatics. https://doi.org/10.1093/bioinformatics/btaa1058
- Jewison T, Su Y, Disfany FM et al (2014) SMPDB 2.0: big improvements to the small molecule pathway database. Nucleic Acids Res. https://doi.org/10.1093/nar/gkt1067
- Dalabira E, Viennas E, Daki E et al (2014) DruGeVar: an online resource triangulating drugs with genes and genomic biomarkers for clinical pharmacogenomics. Public Health Genom. https://doi.org/10.1159/000365895
- Verma J, Luo H, Hu J, Zhang P (2017) DrugPathSeeker: Interactive UI for exploring drug-ADR relation via pathways. In: IEEE Pacific Visualization Symposium
- Jarada T, Rokne J, Alhajj R (2021) SNF-NN: Computational Method To Predict Drug-Disease Interactions Using Similarity Network Fusion and Neural Networks. Res Sq. https://doi.org/10.21203/rs.3.rs-56433/v1
- Cao X, Fan R, Zeng W (2020) DeepDrug: a general graphbased deep learning framework for drug relation prediction. biorxiv. https://doi.org/10.1101/2020.11.09.375626
- Hartenfeller M, Schneider G (2011) Enabling future drug discovery by de novo design. Wiley Interdiscip Rev Comput Mol Sci 1:742–759. https://doi.org/10.1002/wcms.49
- Schneider P, Schneider G (2016) De Novo design at the edge of chaos. J Med Chem 59:4077–4086. https://doi.org/10.1021/acs.jmedchem.5b01849
- Lavecchia A (2015) Machine-learning approaches in drug discovery: methods and applications. Drug Discov Today 20(3):318–331. https://doi.org/10.1016/j.drudis.2014.10.012
- Vyas V, Jain A, Jain A, Gupta A (2008) Virtual screening: a fast tool for drug design. Sci Pharm 76(3):333–360. https://doi.org/10.3797/scipharm.0803-03
- Ertl P, Schuffenhauer A (2009) Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J Cheminform 1:1–11. https://doi.org/10.1186/1758-2946-1-8
- Blaschke T, Olivecrona M, Engkvist O et al (2018) Application of generative autoencoder in De novo molecular design. Mol Inform 37:1–11. https://doi.org/10.1002/minf.201700123
- Jaakkola TS, Haussler D (1999) Exploiting generative models in discriminative classifiers. In: Advances in Neural Information Processing Systems
- Kadurin A, Aliper A, Kazennov A et al (2017) The cornucopia of meaningful leads: applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget 8:10883–10890. https://doi.org/10.18632/oncotarget.14073
- Müller AT, Hiss JA, Schneider G (2018) Recurrent neural network model for constructive peptide design. J Chem Inf Model 58:472–479. https://doi.org/10.1021/acs.jcim.7b00414
- Olivecrona M, Blaschke T, Engkvist O, Chen H (2017) Molecular de-novo design through deep reinforcement learning. J Cheminform 9:1–14. https://doi.org/10.1186/s13321-017-0235-x
- Merk D, Friedrich L, Grisoni F, Schneider G (2018) De novo design of bioactive small molecules by artificial intelligence. Mol Inform 37:3–6. https://doi.org/10.1002/minf.201700153
- Sarkar D (2018) A comprehensive hands-on guide to transfer learning with real-world applications in deep learning. Medium
- Li X, Fourches D (2020) Inductive transfer learning for molecular activity prediction: next-gen QSAR models with MolPMoFiT. J Cheminform. https://doi.org/10.1186/s13321-020-00430-x
- Engkvist O, Norrby PO, Selmi N et al (2018) Computational prediction of chemical reactions: current status and outlook. Drug Discov Today 23:1203–1218. https://doi.org/10.1016/j.drudis.2018.02.014
- Hessler G, Baringhaus KH (2018) Artificial intelligence in drug design. Molecules. https://doi.org/10.3390/molecules23102520
- Domenico A, Nicola G, Daniela T et al (2020) De novo drug design of targeted chemical libraries based on artificial intelligence and pair-based multiobjective optimization. J Chem Inf Model 60:4582–4593. https://doi.org/10.1021/acs.jcim.0c00517
- Ekins S, Puhl AC, Zorn KM et al (2019) Exploiting machine learning for end-to-end drug discovery and development. Nat Mater 18:435–441. https://doi.org/10.1038/s41563-019-0338-z
- Pushpakom S, Iorio F, Eyers PA et al (2018) Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov 18(1):41–58. https://doi.org/10.1038/nrd.2018.168
- Kubick N, Pajares M, Enache I et al (2020) Repurposing Zileuton as a depression drug using an AI and in vitro approach. Molecules. https://doi.org/10.3390/molecules25092155
- Yuan Y, Pei J, Lai L (2020) LigBuilder V3: a multi-target de novo drug design approach. Front Chem 8:1–18. https://doi.org/10.3389/fchem.2020.00142
- Wei L, Wen W, Rao L et al (2020) Cov_FB3D: a de novo covalent drug design protocol integrating the Ba-SAMP strategy and machine-learning-based synthetic tractability evaluation. J Chem Inf Model 60:4388–4402. https://doi.org/10.1021/acs.jcim.9b01197
- Jiménez-Luna J, Grisoni F, Schneider G (2020) Drug discovery with explainable artificial intelligence. Nat Mach Intell 2:573–584. https://doi.org/10.1038/s42256-020-00236-4
- Cavasotto CN, Di Filippo JI (2021) Artificial intelligence in the early stages of drug discovery. Arch Biochem Biophys 698:108730. https://doi.org/10.1016/j.abb.2020.108730
- Wong CH, Siah KW, Lo AW (2019) Estimation of clinical trial success rates and related parameters. Biostatistics 20:273–286. https://doi.org/10.1093/biostatistics/kxx069
- DiMasi JA, Grabowski HG, Hansen RW (2016) Innovation in the pharmaceutical industry: new estimates of R&D costs. J Health Econ 47:20–33. https://doi.org/10.1016/j.jhealeco.2016.01.012
- Ruddigkeit L, Van Deursen R, Blum LC, Reymond JL (2012) Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17. J Chem Inf Model 52:2864–2875. https://doi.org/10.1021/ci300415d
- Mohs RC, Greig NH (2017) Drug discovery and development: role of basic biological research. Alzheimer’s Dement Transl Res Clin Interv 3(4):651–657. https://doi.org/10.1016/2Fj.trci.2017.10.005
- Vamathevan J, Clark D, Czodrowski P et al (2019) Applications of machine learning in drug discovery and development. Nat Rev Drug Discov 18(6):463–477. https://doi.org/10.1038/s41573-019-0024-5
- Niel O, Bastard P (2019) Artificial intelligence in nephrology: core concepts, clinical applications, and perspectives. Am J Kidney Dis. https://doi.org/10.1053/j.ajkd.2019.05.020
- Ahuja AS (2019) The impact of artificial intelligence in medicine on the future role of the physician. PeerJ. https://doi.org/10.7717/peerj.7702
- Rubin EH, Gilliland DG (2012) Drug development and clinical trials-the path to an approved cancer drug. Nat Rev Clin Oncol 9:215–222. https://doi.org/10.1038/nrclinonc.2012.22
- Rautio J, Kumpulainen H, Heimbach T et al (2008) Prodrugs: design and clinical applications. Nat Rev Drug Discov 7(3):255–270. https://doi.org/10.1038/nrd2468
- Harrer S, Shah P, Antony B, Hu J (2019) Artificial intelligence for clinical trial design. Trends Pharmacol Sci 40:577–591. https://doi.org/10.1016/j.tips.2019.05.005
- Fogel DB (2018) Factors associated with clinical trials that fail and opportunities for improving the likelihood of success: a review. Contemp Clin Trials Commun 11:156–164. https://doi.org/10.1016/2Fj.conctc.2018.08.001
- Toh TS, Dondelinger F, Wang D (2019) Looking beyond the hype: applied AI and machine learning in translational medicine. EBioMedicine 47:607–615. https://doi.org/10.1016/j.ebiom.2019.08.027
- Qi Y (2019) Predicting phase 3 clinical trial results by modeling phase 2 clinical trial subject level data using deep learning. Proc Mach Learn Res 106:1–14
- Viceconti M, Henney A, Morley-Fletcher E (2016) In silico clinical trials: how computer simulation will transform the biomedical industry. Int J Clin Trials. https://doi.org/10.18203/2349-3259.ijct20161408
- Magalingam KB, Radhakrishnan A, Ping NS, Haleagrahara N (2018) Current concepts of neurodegenerative mechanisms in Alzheimer’s disease. Biomed Res Int. https://doi.org/10.1155/2018/3740461
- Hussain R, Zubair H, Pursell S, Shahab M (2018) Neurodegenerative diseases: regenerative mechanisms and novel therapeutic approaches. Brain Sci. https://doi.org/10.3390/brainsci8090177
- Levenson RW, Sturm VE, Haase CM (2014) Emotional and behavioral symptoms in neurodegenerative disease: a model for studying the neural bases of psychopathology. Annu Rev Clin Psychol 10:581–606. https://doi.org/10.1146/annurev-clinpsy-032813-153653
- Gitler AD, Dhillon P, Shorter J (2017) Neurodegenerative disease: Models, mechanisms, and a new hope. DMM Dis Model Mech 10:499–502. https://doi.org/10.1242/dmm.030205
- Mak KK, Pichika MR (2019) Artificial intelligence in drug development: present status and future prospects. Drug Discov Today 24(3):773–780. https://doi.org/10.1016/j.drudis.2018.11.014
- Peng J, Guan J, Shang X (2019) Predicting Parkinson’s disease genes based on node2vec and autoencoder. Front Genet 10:1–6. https://doi.org/10.3389/fgene.2019.00226
- Thomas SN, Funk KE, Wan Y et al (2012) Dual modification of Alzheimer’s disease PHF-tau protein by lysine methylation and ubiquitylation: a mass spectrometry approach. Acta Neuropathol. https://doi.org/10.1007/s00401-011-0893-0
- Yousefian-Jazi A, Sung MK, Lee T et al (2020) Functional finemapping of noncoding risk variants in amyotrophic lateral sclerosis utilizing convolutional neural network. Sci Rep 10:1–12. https://doi.org/10.1038/s41598-020-69790-6
- Gupta R, Ambasta RK, Kumar P (2020) Identification of novel class I and class IIb histone deacetylase inhibitor for Alzheimer’s disease therapeutics. Life Sci. https://doi.org/10.1016/j.lfs.2020.117912
- Jamal S, Grover A, Grover S (2019) Machine learning from molecular dynamics trajectories to predict caspase-8 Inhibitors against Alzheimer’s disease. Front Pharmacol 10:1–13. https://doi.org/10.3389/fphar.2019.00780
- Chen HY, Chen JQ, Li JY et al (2019) Deep learning and random forest approach for finding the optimal traditional Chinese medicine formula for treatment of Alzheimer’s Disease. J Chem Inf Model 59:1605–1623. https://doi.org/10.1021/acs.jcim.9b00041
- Ponzoni I, Sebastián-Pérez V, Martínez MJ et al (2019) QSAR Classification models for predicting the activity of inhibitors of beta-secretase (BACE1) associated with Alzheimer’s disease. Sci Rep 9:1–13. https://doi.org/10.1038/s41598-019-45522-3
- Kaiser TM, Dentmon ZW, Dalloul CE et al (2020) Accelerated discovery of novel Ponatinib Analogs with improved properties for the treatment of Parkinson’s disease. ACS Med Chem Lett 11:491–496. https://doi.org/10.1021/acsmedchemlett.9b00612
- Shao YM, Ma X, Paira P et al (2018) Discovery of indolylpiperazinylpyrimidines with dual-target profiles at adenosine A2A and dopamine D2 receptors for Parkinson’s disease treatment. PLoS ONE 13:1–27. https://doi.org/10.1371/journal.pone.0188212
- Chen ZD, Zhao L, Chen HY et al (2020) A novel artificial intelligence protocol to investigate potential leads for Parkinson’s disease. RSC Adv 10:22939–22958. https://doi.org/10.1039/d0ra04028b
- Deng L, Zhong W, Zhao L et al (2020) Artificial intelligencebased application to explore inhibitors of neurodegenerative diseases. Front Neurorobot. https://doi.org/10.3389/fnbot.2020.617327
- Oh M, Ahn J, Yoon Y (2014) A network-based classification model for deriving novel drug-disease associations and assessing their molecular actions. PLoS ONE 9:1–12. https://doi.org/10.1371/journal.pone.0111668
- Zhu Y, Jung W, Wang F, Che C (2020) Drug repurposing against Parkinson’s disease by text mining the scientific literature. Libr Hi Tech 38:741–750. https://doi.org/10.1108/LHT-08-2019-0170
- Vatansever S, Schlessinger A, Wacker D, et al (2020) Artificial intelligence and machine learning-aided drug discovery in central nervous system diseases: state-of-the-arts and future directions. Med Res Rev Online ahead of print
- Stokes JM, Yang K, Swanson K et al (2020) A deep learning approach to antibiotic discovery. Cell. https://doi.org/10.1016/j.cell.2020.01.021
- Mamoshina P, Vieira A, Putin E, Zhavoronkov A (2016) Applications of deep learning in biomedicine. Mol Pharm 13(5):1445–1454. https://doi.org/10.1021/acs.molpharmaceut.5b00982
- Preuer K, Klambauer G, Rippmann F, et al (2019) Interpretable deep learning in drug discovery. In: Lecture Notes in Computer Science (including subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics)
- Ramsundar B, Liu B, Wu Z et al (2017) Is multitask deep learning practical for pharma? J Chem Inf Model. https://doi.org/10.1021/acs.jcim.7b00146
- Grace K, Salvatier J, Dafoe A, et al (2017) When Will AI Exceed Human Performance? evidence from AI experts. J Artif Intell Res 62:1–48 https://arxiv.org/abs/1705.08807
- Altae-Tran H, Ramsundar B, Pappu AS, Pande V (2017) Low data drug discovery with one-shot learning. ACS Cent Sci 3:283–293. https://doi.org/10.1021/acscentsci.6b00367
文章出处登录后可见!