AI after AlphaFold
新的预印本描述了一种新的无参数原子坐标几何变换器,用于预测蛋白质中的生物界面
它运行速度如此之快,甚至可以扫描大量蛋白质结构以搜索易于相互作用的氨基酸。
机器学习、人工神经网络和其他基于“人工智能”的数学方法(我不太喜欢这个词,但它坚持!)几十年来一直应用于科学问题。但众所周知,它们现在有着前所未有的应用,以激进的方式改变着化学和生物学等科学。
现代人工智能最引人注目的应用之一可能是预测蛋白质结构,它始于大约 5 到 10 年前,并在 2020 年末和 2021 年推出的 AlphaFold 2 成为罪魁祸首。我在之前的文章中详细讨论了 AlphaFold 2:
从事蛋白质结构预测的科学家们首先对 AlphaFold 的影响感到负面情绪,因为它有点扰乱了他们自己的领域,实现了他们许多期待已久的目标。然而,在短暂的哀悼之后,他们接受并实际上利用了 AlphaFold 来发现新发现并开发新工具,其中许多我已经讨论过:
毕竟,AlphaFold 2 并没有解决分子和结构生物学中的所有相关问题。事实上,它只解决了这个巨大难题的一小部分(这根本不会让它变小!)AlphaFold 2 解决了什么(我说“有点”,因为即使这个问题也没有完全解决;而且我继续强调 AlphaFold 2,因为它的第一个版本还不太擅长)正在预测蛋白质的所谓“三级结构”,这本质上意味着它们的组成原子如何在 3D 空间中排列。
但是蛋白质结构有几个层次的复杂性。蛋白质是氨基酸的长线性链,可折叠成 3D 结构以实现三级结构,但这些又可以形成高级结构,即多个蛋白质之间或蛋白质与其他生物大分子(如核酸(DNA 和 RNA)或与膜、离子、小分子等。事实上,在大多数情况下,蛋白质的生物学功能存在于或被这些复合物以生理相关的方式调节。
当一种蛋白质与另一种蛋白质相互作用时,我们谈论的是蛋白质-蛋白质复合物,AlphaFold 2 可以预测其中的一些相互作用(尤其是在其“AlphaFold Multimer”风味中),但它还不是很擅长。如果我们考虑蛋白质可以建立的其他类型的相互作用,AlphaFold 就出局了。它只是不旨在预测蛋白质与除蛋白质以外的分子之间的相互作用,例如 DNA、RNA、离子、氨基酸等小分子、代谢中间体、细胞信号分子等或生物膜及其成分、脂质。
对这些其他相互作用进行建模是在原子水平上对生物结构、相互作用和功能进行建模的下一步,许多团队多年来一直致力于此工作。如果 Deepmind 本身现在继续解决蛋白质可以参与的其他一些相互作用,我不会感到惊讶。特别是,小分子结合的具体预测与制药有很大的相关性,因为大多数临床使用的化合物本身就是与特定蛋白质相互作用的小分子。
要了解更多关于人工智能在蛋白质结构预测和结构生物学/结构生物信息学中的下一条路线,您可以查看我最近写的这篇文章:
使用无参数几何变换器预测蛋白质将与什么相互作用
我工作的实验室的一个新预印本现在使用一种新的公式解决了这个确切的问题:
给定蛋白质的结构或模型,预测它可以形成什么界面来结合其他蛋白质、核酸、脂质、离子或其他种类的小分子。
领导这项工作的博士生开发了一种几何转换器,可以读取和处理输入蛋白质的 3D 坐标,并产生残基特异性分数,预测蛋白质的每个氨基酸成为与其他蛋白质界面的一部分的可能性,使用核酸/s,使用离子/s 等。该方法在蛋白质结构转换器之后称为 PeSTo,具有非常高的准确性,几乎不会混淆界面,并且与替代方法相比有几个非常有利的点:
- 运行该模型不涉及对输入蛋白质表面的任何计算,因为大多数替代方法都需要。表面计算的计算速度很慢,并且对 3D 结构中的错误非常敏感。
- 该模型以毫秒为单位运行,包括其加载时间,这意味着您可以在短时间内处理大量结构。事实上,它是如此之快,以至于它可以在几秒钟内处理整个分子动力学轨迹,这对于识别只有在蛋白质移动时才能访问的瞬态界面很有用,正如我们所展示的。我们还可以处理整个人类蛋白质组,发现新的生物学。
- 该模型不依赖任何参数化甚至分类,因为它完全基于原子元素和空间位置进行训练。因此,尽管我们将 PeSTo 应用于蛋白质及其 C、N、O 原子,但它应该很容易重新训练用于其他目的,例如在材料科学中。
一种新的原子坐标几何变换器
让我提几个有关 PeSTo 工作原理的关键点。更多详情,可以参考预印本:
PeSTo 将蛋白质结构视为点原子云,通过保证平移不变性的成对距离和相对位移矢量来表示几何结构。每个点原子仅使用其元素名称来描述,而没有其他方法使用的数值参数化,例如半径或电荷。每个原子通过几何变换器编码,该变换器通过标量和矢量状态以及从周围原子在增加距离处计算的距离来解释其局部邻域。根据查询,该描述符通过网络传播,通过多头注意力操作产生特定于原子的输出。然后通过两个附加模块为每个蛋白质残基收集基于原子的输出,这些模块最终预测蛋白质的每个残基是否可能位于界面处。
基于来自蛋白质数据库的数据集,我们训练模型以输出参与蛋白质-蛋白质、蛋白质-核酸、蛋白质-离子、蛋白质-配体、蛋白质-离子和蛋白质-脂质界面的残基概率.
Webserver 的实现和一个具体的例子
预印本包括一些选定的示例。我将在这里向您展示我在 https://pesto.epfl.ch 的网络服务器实现上运行的一个具体示例[0]
当您访问该网站时,您可以选择对以下内容进行预测:
- 来自 PDB 的蛋白质结构,使用 4 个字符的 ID 输入
- 在 AlphaFold-EBI 的数据库中预先计算的蛋白质模型,作为 UniProt ID 输入
- 您上传的蛋白质结构/模型。
让我们在这里尝试一下 PDB 的结构,因为这允许我介绍输入页面的另一个功能:
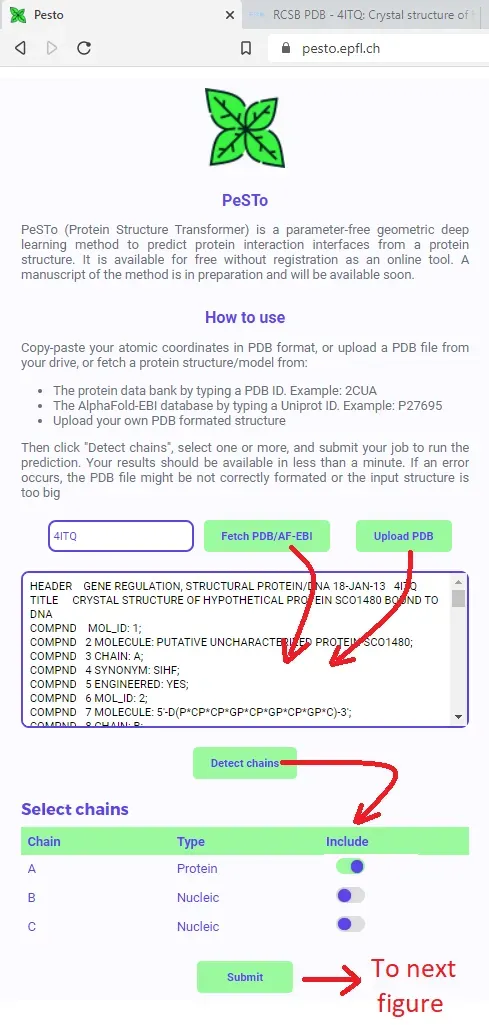
我故意服用了4ITQ。这是与 DNA 结合的蛋白质的 X 射线结构。 PDB 中注释的生物组装标记了一个特定的蛋白质-DNA 表面,但我为另一项工作对相关蛋白质进行的基于溶液的 NMR 实验揭示了更广泛的 DNA 相互作用表面。 PeSTo 预测什么?
让我们先看看 PeSTO 对这种蛋白质的所有预测:

PeSTo 会产生一个相当大的残基表面,这些残基可能参与结合核酸,还有一个环,可能是结合离子。没有任何东西可以结合除离子以外的其他蛋白质、脂质或配体。
DNA 结合的界面非常大,与我之前论文中的 NMR 结果非常一致,这表明通过至少两个界面进行结合,而不仅仅是 X 射线结构所提出的一个界面。此外,该论文中的 AFM 实验表明,这种蛋白质在 DNA 中引入了强大的环和扭结,暂时是因为它迫使 DNA 缠绕在它周围,展开它以实现一些尚不清楚的生物学功能。
在分子动力学模拟和折叠体中发现界面的应用
PeSTo 运行得如此之快,以至于我们可以将其应用于大量结构。对于我们的预印本,我们尝试将其作为一种工具来识别受分子模拟影响的蛋白质界面,以及收集完整人类蛋白质组的结构。
应用于分子动力学模拟 PeSTo 非常有用,因为它可以自动检测在用于开始模拟的结构中可能不明显但可能在动力学中暴露出来的界面。这对于发现所谓的神秘口袋尤其有效,即蛋白质表面的小口袋随着蛋白质的移动而出现和消失,因此可能会在静态 X 射线结构中丢失。
进一步和相关阅读
The preprint:
一个连接到我所有关于 AlphaFold 和蛋白质结构预测的文章的中心故事:
Deepmind 也致力于将 AI 应用于其他科学领域:
一种预测蛋白质相互作用但通过表面上的几何深度学习的方法:
www.lucianoabriata.com 我撰写和拍摄我广泛兴趣范围内的所有内容:自然、科学、技术、编程等。成为 Medium 会员以访问其所有故事(平台的附属链接)没有成本的收入)并订阅以通过电子邮件获取我的新故事。要咨询小型工作,请在此处查看我的服务页面。你可以在这里联系我。[0][1][2][3][4]
文章出处登录后可见!